Biomed Res Bull. 2(4):188-198.
doi: 10.34172/biomedrb.2024.26
Review Article
A Comprehensive Insight Into the Role of Valproic Acid in Neurodegenerative Disorders: An Updated Review
Hesameddin Razilou 1  , Seyyed Sina Hejazian 1, Shahed Meshkini 1, *
, Seyyed Sina Hejazian 1, Shahed Meshkini 1, *
Author information:
1Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
Degenerative nerve diseases are caused by the progressive loss of the structure and function of neurons. Numerous signaling pathways have been suggested to be involved in neurodegenerative disease. Valproic acid is a branched-chain fatty acid used to treat bipolar disorder and epilepsy. Valproic acid modulates immunity by the activation of monocyte-derived macrophages. The present review study focused on the role of Valproic acid in neurodegenerative diseases such as MS and stroke.
Keywords: Sodium valproate, Neurodegenerative diseases
Copyright and License Information
© 2024 The Author(s).
This is an open access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Funding Statement
No Funders.
Introduction
Sodium Valproate
Sodium valproate (VPA, 2-Propylpentanoic acid) is a first-line broad-spectrum anticonvulsant drug with mood-stabilizing properties, which is widely used in the treatment of convulsion and bipolar diseases. VPA is a short-chain (pentameric) 8-carbon fatty acid that was first obtained from Valeriana officinalis and is used worldwide.1 Multiple mechanisms of action have been proposed for VPA, including sodium channel blockade, decreased γ-aminobutyric acid transaminase (GABA-T) activity, increased extracellular GABA neurotransmitter content, decreased succinate semialdehyde dehydrogenase (SSADH), and effect on the T-type calcium channel.2,3 Common side effects of VPA in therapeutic doses include nausea, vomiting, gastrointestinal upset, and heartburn. In addition, tremor may be seen at higher doses of the drug. Its rare but deadly complication is hepatotoxicity that occurs idiosyncratically and manifests itself with increased liver enzymes. It also alters the metabolism of several drugs, which limits its use.4,5
Neurodegenerative Diseases
Neurodegenerative diseases are age-related diseases that directly affect the central nervous system and include a wide range of diseases, the most common of which include Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and multiple sclerosis (MS). Numerous mechanisms have been suggested for the pathogenesis of these diseases and include the involvement of both neuronal and non-neuronal cells of the brain, such as the glial cells. They cause various clinical symptoms, including movement, speech, memory, and cognitive impairment.6,7 Given that the prevalence of these diseases is rapidly increasing due to the increase in the average age of the population and the growth in the number of cases in developed countries, the need for proper treatment has increased considerably. However, in most of the cases, there is no definitive and appropriate treatment. Therefore, the use of alternative drugs to treat these patients has become more important than ever. Among the drugs that have recently been suggested to be effective in neurodegenerative diseases is VPA.8-11
Valproic Acid and Neuroprotection
The production of new drugs requires lots of money and time. The proposed solution to this problem is to use the unknown properties of existing drugs. VPA is one of the well-known drugs for treating central nervous system diseases. New therapeutic effects in favor of its potential in treating neurodegenerative diseases have been discovered. These effects include neuroprotective effects on dopaminergic neurons by several mechanisms, including increased expression of glial cell line-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF) by inhibiting histone deacetylase (HDAC) activity and anti-inflammatory effects in neurons by attenuating release of pro-inflammatory factors from microglia.12-14 It also affects enzymatic pathways of extracellular-signal-regulated kinases (ERK), phosphatidylinositol 3 kinase/akt-1, and glycogen synthase kinase 3B (GSK-3B).15 In addition, it has neuroprotective effects against glutamate excitotoxicity, which is mediated by increased alpha-synuclein expression following inhibition of HDAC and increased Bcl-2 expression.16 A study showed that VPA has anti-inflammatory and neuroprotective effects in mice with ischemic stroke.17 VPA has also been shown to prevent the death of hippocampal cells following ischemic inflammation.18 Another mechanism suggested for the neuroprotective properties of this drug is the inhibition of TNF-α release and NO production from microglia cells.19 This drug is even effective in inhibiting the activity of Tcells, which are involved in nerve damage.20 Besides, several other mechanisms and multiple regulatory pathways are involved in the neuroprotective properties of this drug.21,22 In the following, we will review the studies that have investigated the role of this drug in neurodegenerative diseases.
Valproic Acid and Alzheimer’s Disease
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia in the elderly, leading to progressive loss of memory, as well as cognitive and behavioral impairments. AD is an irreversible neurodegenerative disease and several mechanisms are involved in its pathogenesis, the most prominent of which are extracellular accumulation of senile plaques (SP), mainly composed of amyloid beta (Aβ), and intracellular neurofibrillary tangle (NFT), mainly composed of hyperphosphorylated Tau protein.23 Aβ is the result of proteolytic cleavage of amyloid precursor protein (APP) and hyperphosphorylation of tau protein is the result of dysfunction of several kinases, including GSK-3B, which is associated with impaired axonal transport.24 In addition, studies have shown cholinergic system impairments in mice models of AD.25 It is estimated that 51.6 million people worldwide suffer from AD, although gender differences in the prevalence and course of the disease have been reported.26 Other common symptoms in AD patients include behavioral and psychological symptoms of dementia (BPSD), which are accompanied by agitation, violence, inappropriate sexual behavior, delusion, and misidentification.27 The most common treatment for BPSD is antipsychotic drugs, especially atypical antipsychotic drugs; however, there are concerns about their side effects.28
Use of Valproic Acid in Alzheimer’s Disease
It has been shown that HDAC inhibitors, including VPA, improve spatial learning and memory and cognitive impairments in AD patients; however, VPA does not improve memory in wild-type (WT) mice.29 Numerous animal and human studies have evaluated the efficacy of VPA in the treatment of AD. The study conducted by Yao et al investigated the efficacy of VPA in male and female APPswe/PS1dE9 mice as mice models of AD.30 The use of VPA in these mice (100 mg/kg/d for 3 months intraperitoneally) improved long-term recognition memory, spatial learning, and memory. HDAC activity in the hippocampus of these mice was increased by 96% compared to the WT and vehicle mice, which is in accordance with increased acetylation of H3 lysine 9 and H4 lysin 8 by 28% and 32%, respectively.31 Considering the role of HDAC2 in increasing the number of neurons and synapses, HDAC2 level was further investigated in this study. No significant differences in HDAC2 expression were observed between study groups; however, the activity level of this enzyme was elevated among VPA-treated mice.30,32 The other mechanism of action introduced in this study for VPA was the increase in the expressions of synaptic plasticity-associated proteins including CRB, CAMKII, and CAM in the hippocampus, which are involved in improving spatial learning and memory.30 In another study that used higher doses of VPA for a shorter period of time (200 mg/kg/d for 2-3 weeks), similar effects were observed in the long-term memory of mice with hypoxia-induced memory deficit.33 Even low doses of VPA for 4 weeks (30 mg/kg/d) have been reported to be effective in improving spatial learning and memory impairment in mouse models of AD.34,35
Studies have shown the neuroprotective role of VPA in AD. Xuan et al reported that the number of NeuN + cells in the hippocampus and ChAT-immunoreactive neurons in the vertical diagonal band of Broca (VDB) and medial septum in APP/PS1 mice increased after treatment with VPA.36,37 It was also shown that the activity of microglia and astrocytes, which are normally seen around Aβ plaques, decreases in these mice, resulting in reduced levels of IL-1β and TNFα in the hippocampus and cortex.38 In addition, the study by Xuan et al showed reduced levels of cytokines and cytotoxic molecules in VPA-treated mice.39
Both Aβ deposition and soluble form of Aβ are associated with cognitive impairments in AD.40 VPA has been shown to be effective in the reduction of both forms of Aβ in transgenic mice. Interestingly, it has been shown that Aβ plaques stimulate the production of inflammatory cytokines by microglia, and these cytokines accelerate the formation of Aβ plaques in AD.41,42 Therefore, it can be concluded that VPA prevents further damage to the brain in AD through both mechanisms of reducing the production of cytokines and the formation of Aβ plaques. VPA increases the expression and activity of BCL-2 as a protein involved in neuroprotection, resulting in anti-apoptotic effects of this drug in mouse models of AD.42 It has been also reported that VPA inhibits the GSK-3B activity through phosphorylation at serin-9 (producing phospho-GSK-3B-ser9), which is associated with reduced levels of Aβ.43 Phosphorylation of NFkB-p65 around the Aβ plaques causes inflammation. VPA can reverse this process, resulting in neuroprotective effects.44,45
As mentioned before, gender differences are seen in AD. These differences are seen in VPA-treated mice as well. The study by Long et al showed that the improvement of most signs of AD by VPA treatment was greater in male mice than in female mice.46 In addition, Aβ plaque, Aβ40 and Aβ42 reduction, and improvement in synaptic activity and structure was greater in male mice than in female mice. Moreover, the Aβ cleavage by an increase in neprilysin was greater in male mice. No significant differences were seen in presynaptic vesicle density between the two genders.47 It has been shown that melatonin has a great role in Aβ reduction and Tau protein hyperphosphorylation, which may improve synaptic plasticity and cognitive performance in aged mice.48 Circulating melatonin levels and expression of its receptor, M2, in the hippocampus of AD patients are reduced even in preclinical stages.49 Hong et al reported that M2 levels in CA1, CA2, CA3, and DG regions of hippocampus in Sprague-Dawley rats increased after 17 days of treatment with VPA. Besides, the efficacy of VPA in different stages of AD was assessed which showed a greater reduction in neurite plaque formation in the early stages of the disease. In addition, they reported that the impact of VPA lasted for about two months after discontinuation.34
In a clinical study, the efficacy of donepezil + VPA in AD patients with BPSD was compared to donepezil + quetiapine, indicating no differences. Both therapies had satisfactory results; however, the first combination therapy was more cost-effective than the other. The same study showed that VEGF increased after VPA treatment, especially in patients with excellent prognosis.50 Herrmann et al evaluated the efficacy of VPA in elderly patients with different stages of AD who suffered from aggression and agitation. The patients were treated with an average dose of 1135 mg/d for about 6 weeks. VPA therapy did not alleviate the symptoms, but rather it intensified them.51 Many studies have used divalproex, a drug similar to VPA, in the treatment of aggressive behaviors in AD patients, which showed no favorable results.52
Valproic Acid and Parkinson’s Disease
Parkinson’s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disease and the most common movement disorder associated with tremor, stiffness, bradykinesia, and postural instability. Non-motor symptoms in this disease include autonomic disorders, cognitive impairment, and sleep disorders. The main pathological change in this disease is the destruction of dopaminergic cells in the nigrostriatal pathway. The diagnostic hallmark of these cells is the presence of intracytoplasmic inclusions called Lewy bodies (LB) containing α-synuclein (αSyn) and Lewy neurites.53,54 The exact mechanism of this disease is not known; however, epigenetic disorders such as DNA methylation or Histon remodeling have been found to be effective in the pathogenesis of PD.55 Mutant αSyn in PD patients can promote the translocation of methyltransferase 1 into the cytoplasm, causing hypomethylation of the promoter region of the αSyn gene (SNCA). In addition, the αSyn has been shown to mask histone proteins and thus prevent their acetylation, leading to hypoacetylation, which can ultimately induce apoptosis.56,57 There is no definitive cure for this disease; however, treatments may alleviate symptoms and prevent its progression. However, levodopa and dopaminergic drugs have been approved.58 PD is a progressive disease and patients’ need for dopamine increases over time which exposes patients to side effects such as on-off fluctuations, dyskinesias, dystonia, psychosis, and/or behavioral syndromes such as impulse control disorders (ICDs). ICDs include unrestrained gambling, hypersexuality, compulsive shopping, compulsive eating, hobbyism, punding, and compulsive medication use.59,60
Use of Valproic Acid in Parkinson’s Disease
Many animal studies have evaluated the efficacy of VPA in the treatment of mice models of PD. It has been shown that pretreatment with VPA in PD-induced mice using Rotenone and 6-hydroxydopamine shows neuroprotective effects on striatal dopaminergic neuronal terminals and dopaminergic cell bodies in substantia nigra pars compacta (SNpc). However, PD induction using Rotenone and 6-hydroxydopamine is not based on protein deposition, which is the main pathological mechanism in patients with PD.61,62 In another study,the same neuroprotective properties of VPA were shown in PD-induced mice by the mitochondrial toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).63 It is important to note that in mouse models of PD, by the time of symptoms emergence, 60%-70% of SNpc neurons are destroyed and striatal dopamine levels are reduced by up to 80%. Therefore, the use of VPA before the induction of PD in mice may not mimic the exact clinical condition of PD patients; therefore, the results may not be reliable.64 In another study by Harrison et al, the neuroprotective properties of VPA were investigated in mice after induction of PD using the neurotoxin Lactacystin (a proteasome inhibitor). This study not only demonstrated that VPAprotects neurons against neurodegeneration but also showed that VPA reverses the degeneration of dopaminergic nigrostriatal neurons in these mice. These effects were not limited to nigrostriatal cells and were observed in other brain regions as well. MRIwas also used to evaluate neurological change, which showed a decrease in midbrain volume and an increase in ventricular volume following induction of PD, both of which were attenuated in VPA-treated mice. In this study, the levels of BCL2 as an anti-apoptotic factor and BAD as a pre-apoptotic factor were examined, the first of which increased after treatment with VPA and the second decreased.65
Studies have shown an increase in neurotrophic factors, BDNF and GDNF, and acetylation of histones in mouse models of PD following VPA use. VPA has also been shown to be effective in alleviating movement disorders in these mice.66,67 In a study by Carriere et al, unilateral intrastriatal rotenone injection not only caused movement disorders contralateral to the injection side but also led to an increase in ipsilateral forelimb use, both of which were reversed by VPA.68 VPA has also been shown to be effective in the alleviation of non-motor symptoms such as cognitive and emotional deficits in mouse models of PD.69 In one of these studies, Castro et al reported significantly increased levels of striatal dopamine following VPA use in mouse models of PD.70 Immunohistochemical studies have shown partial and complete maintenance of tyrosine hydroxylase immunoreactivity in striatum and substantia nigra of PD-induced mice, respectively.71
The prevalence of pathological gambling, which is defined as excessive uncontrolled gambling despite financial losses and social problems, varies from 3.4% to 6.1% among PD patients.69 A case report study by Degirmenci and Kececi reported successful treatment of pathological gambling in a 52-year-old man who had PD for 3 years. The patient in this study had symptoms of inappropriate behaviors such as compulsive shopping, unreasonable selling, and pathological gambling, which had gradually worsened. The patient also suffered terrifying visual hallucinations at night and was treated with levodopa and several other drugs. Assuming the symptoms were side effects of the drugs, their dose was reduced, resulting in alleviation of the sign; however, the pathological gambling remained uncontrolled and treatment with VPA was started for the patient (500 mg/d). In the second week of treatment, the patient’s symptoms began to decrease and after 1 month of treatment, they completely disappeared. No side effects after treatment with VPA were seen.72
Dopamine dysregulation syndrome (DDS), also called hedonistic homeostatic dysregulation syndrome, is defined as excessive consumption of dopaminergic drugs by PD patients, which is seen in 1%-2% of these patients.73,74 In a study by Sriram et al, 4 cases of DDS were treated with VPA which alleviated their excessive desire for dopaminergic use after a short period of therapy.75 In addition, in a case series study by Hicks et al, VPA was used to treat ICD symptoms in three patients with PD, which showed satisfactory results. These patients suffered from hypersexuality, episodes of extreme excitement, and compulsive behaviors and showed signs of DDS. Treatment with VPA reduced inappropriate behaviors of all patients; therefore, the dose of needed levodopa was reduced. In two patients, VPA was discontinued after a period of time, leading to the return of ICD symptoms.76,77
VPA has been evaluated in the treatment of PD patients and the results showed that the number of on-off fluctuations increased in patients.78 In another study, Ximenes et al showed that the levels of OX-42 and GFAP (reactive factors of microglia and astrocytes) increased following induction of PDin mice with 6-OHDA, respectively, which were both reversed by the administration of VPA. TNF-α and HDAC levels also decreased following VPA treatment. All of these changes were seen in the CA1 and CA3 regions of the hippocampus and the cortex.79
In addition to all these studies, it should be noted that VPA is known as a drug with the potential to induce PD.80 In a case report of a 67-year-old male patient with PD, the patient’s symptoms worsened following VPA use. In this study, following the aggravation of the patient’s cognitive and psychological symptoms, rivastigmine (3 mg/d) and VPA (500 mg twice a day) were started, which led to severe exacerbation of movement disorders in the patient to the point he was unable to walk without assistance. Cessation of the drug significantly alleviated the movement disorder of the patient after a week. Symptoms completely disappeared after two months. However, the patient’s cognitive and psychological disorders returned rapidly 81.
Valproic Acid and Multiple Sclerosis
Multiple Sclerosis
MS is the most frequent chronic inflammatory, demyelinating, and neurodegenerative disease of the central nervous system, which is the most common cause of non-traumatic neurological disability in young people. Multiple mechanisms are involved in the pathogenesis of this disease, including oxidative stress, excitotoxicity, autoimmunity, and hormonal disorders. The main cells involved in the pathogenesis of the disease are TH1 and TH17 cells.82,83 The disease typically occurs between the ages of 25 and 45 and presents with a variety of symptoms, all of which are somehow related to the central nervous system.84 Numerous attempts have been made to find an effective treatment for the disease; however, no definitive cure has been found to date, and only ocrelizumab and Siponimod among the 17 FDA-approved drugs are relatively effective in reducing the debilitating symptoms of the disease.85-87 In addition, the current treatments are most effective in the relapsing-remitting form and not the progressive form of the disease. Therefore, there is an urgent need for new therapeutic ways for this disease.88
Use of Valproic Acid in MS
Numerous studies have attempted to find new therapeutic strategies for the treatment of MS.89-91 VPA is among the drugs that have been reported to be useful in the treatment of this disease, which is not unexpected due to its neuroprotective effects.92,93 The studies have shown the efficacy of VPA in the treatment of MS by alleviating the symptoms of patients.94,95 The most recent two studies on the use of VPA in the treatment of MS patients were performed in 2015, the results of which are inconsistent with each other. In one of these studies, which was a case report study, the effectiveness of VPA in the treatment of pseudobulbar affect in MS patients was evaluated.93 Pseudobulbar affect is a disabling feature of unknown pathophysiology found in many people with central nervous system diseases and manifests itself with sudden episodes of laughter and crying.94 The patient treated in this study suffered from incessant crying that was completely alleviated after three consecutive days of treatment with VPA. However, following a change in the patient’s medication regimen to an FDA-approved drug, Nuedexta (dextromethorphan plus quinidine) at discharge, the patient’s symptoms returned completely after 6 days, forcing physicians to repeat the course of VPA treatment. Similar to the first course, the patient’s symptoms disappeared quickly. The results of this study were in contrast to another study conducted in the same year by Nielsen et al.95 In this large retrospective cohort, the risk of developing MS in people of different races using VPA was evaluated for the first time. They found that VPA did not prevent MS from occurring, but rather it increased the risk of developing MS in patients with a previous history of VPA use. The risk was higher in the first year after stopping the drug, and it was no longer observed after five years. This probably occurred in the context of the reversal of the neuroprotective effects of the VPA following its cessation. Therefore, unlike the previous three studies, the finding of this study was not in favor of the therapeutic effects of VPA in MS patients.95
In addition to human studies, several animal studies have examined the impact of VPA on MS. 90 Optic neuritis is the inflammation of the optic nerve that occurs in many patients with MS, and it may be the first manifestation of their disease, which occurs in 20% of cases.89 It has been shown that the use of VPA reduces inflammation in the spinal cord and optic nerve and therefore is effective in reducing demyelination and degeneration in MS patients. The ASK1 pathway has been shown to regulate the production of chemokines in astrocytes and the progression of demyelination through the production of pro-inflammatory factors.80 In a study, in addition to VPA, ASK1 pathway inhibitors were used to treat EAE mice, which showed synergistic effects with VPA. The mechanism proposed in this study for the neuroprotective effects of VPA in MS was to reduce the infiltration of T cells into the optic nerve and spinal cord and to suppress the activity of glial cells, the latter of which may itself be due to T-cell suppression.
VPA suppresses degeneration and demyelination in the optic nerve in MS models. Additionally, it has been reported to be effective in inducing remyelination and thus improving the neural function of these mice. In two separate studies were shown that the number of myelinated neurons increased in mouse models of MS following VPA treatment.91,92 In the same studies, VPA was also shown to be effective in increasing neural stem cells and oligodendrocyte precursors in the brain, which in turn can be useful in reversing neurological damage in MS patients.85,86 For the first time, Rossi et al used a combination of two drugs, VPA and α-linolenic acid, in mice models of MS, the results of which were satisfactory.87 In addition to the efficacy of VPA in preventing or reversing pathological changes, numerous studies have demonstrated improvements in the performance of MS mouse models. In a study performed on Dark Agouti (DA) mice with EAE, a combination of VPA and T4 was used for three consecutive days in these mice, which significantly reduced the symptoms and pathology of the rat brain. This study showed that VPA has immunomodulatory properties and reduces the production of oligodendrocyte progenitors. This study is of great importance because it showed that VPA can be therapeutically effective at low doses.90
Valproic Acid and Huntington’s Disease
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant genetic disorder that occurs due to the replication of the three-nucleotide (cytosine-adenine-guanine) sequence in the Huntington (HTT, OMIM 613004) gene located on chromosome 4, causing corticospinal involvement as cerebral cortex atrophy.89 The importance of the disease is primarily due to its progressive and consequently fatal nature within 15 to 20 years from the onset; otherwise, the prevalence of the disease is low and reaches its maximum at 16 cases per million population.92 The symptoms of the disease are more common in middle-aged people and include chorea, dystonia, cognitive disorders, and psychological changes, the last two of which usually occur before movement disorders.95 Common treatments for this condition include using neuroleptic antidopaminergic (such as haloperidol and olanzapine) or dopamine-lowering drugs (such as tetrabenazine), long-term use of which has always been accompanied by concerns due to their side effects such as extrapyramidal symptoms, difficulty swallowing, and bradykinesia.88 Therefore, the use of alternative therapies has received more and more attention and many studies have evaluated the efficacy of different drugs in the treatment of these patients, among which VPA has shown promising effects due to its neuroprotective properties.90
Use of Valproic Acid in Huntington’s Disease
Among the pathogenic mechanisms of the HD disease, excitotoxicity, decreased medium-sized GABAergic neurons in the striatum, and increased histone deacetylation have been studied,90 the three mechanisms shown to be modulated by VPA.89 The first study to evaluate the effect of VPA in HD was conducted by Tan et al in 1976, in which the efficacy of VPA and L-Dopa in the treatment of a female patient with HD was compared. It was shown that despite the effectiveness of VPA in alleviating the disease, the effect of the drug decreased after a few weeks, whereas no such change in efficacy was observed in treatment with L-Dopa.67 Other clinical studies evaluating the efficacy of VPA in the treatment of HD were performed, in one of which the efficacy of VPA in reducing myoclonus of HD patients was assessed.88 Action myoclonus or myoclonic hyperkinesia is one of the rare but debilitating manifestations in HD patients that may be confused with chorea.91 Studies have shown the role of the GABA neurotransmitter in the development of myoclonus in HD patients. In this regard, VPA may have beneficial effects on the myoclonus as an extracellular GABA enhancer.92 In this study, 8 HD patients with a previous history of myoclonic hyperkinesia were treated with different doses of VPA. Only one of the patients who was treated with a lower dose of VPA (300 mg/d) did not respond well to the treatment and higher doses of the drug were significantly effective on patient’s symptoms. Even a patient with a previous history of VPA use (1050 mg/d) experienced reduced symptoms by increasing the dose of VPA to 1800 mg/d. All of this evidence suggests a dose-dependent effect of VPA on HD-induced myoclonus.93 Clinical studies investigated the combined effect of VPA and olanzapine in HDpatients.94 Two 39- and 52-year-old patients with a history of HD who were treated with Haloperidol and recently had worsening symptoms were admitted to the hospital. They underwent a combination therapy of VPA and Olanzapine (5 mg/d and 1500 mg/d, respectively). They showed significant improvement in symptoms of movement disorders, psychosis, and aggression. However, recovery in these patients took 7-8 weeks. Due to the lack of response to Haloperidol as the standard treatment for HD and the satisfactory results of VPA + Olanzapine use in this study, further investigations of this combination therapy seem reasonable.86 The most recent study on the use of VPA in the treatment of HD was the study conducted by Whiting et al in 2018. In this study, they evaluated the efficacy of VPA in the treatment of behavioral disorders in a 59-year-old female patient with an 8-year history of Huntington’s disease. The patient suffered from relatively severe choreoathetosis and dysarthria and was hospitalized due to the intensification of aggressive behaviors since a month ago. After initial treatment and observing no improvement, the administration of VPA was started for the patient and a blood level of 95 µg/mL was achieved within 4 days. The patient responded quickly to treatment within a week of treatment, and his aggressive behaviors were significantly reduced. The patient was followed up for only one month when no recurrence of symptoms was observed. However, further studies are needed to evaluate the long-term effectiveness of the treatment.87
In addition to numerous clinical studies, several animal studies have been performed to evaluate the effect of VPA in HD, all of which have shown good efficacy. Among them recent study was performed on N171-82Q mice as a mouse model of HD.88 In a study, the efficacy of VPA in preventing HD in mice was evaluated and it was shown that the onset of the disease was delayed in mice treated with VPA; therefore, their survival was improved by 31.4% which was higher than the results obtained in another study that examined the efficacy of phenylbutyrate on survival of mice models of HD.89 In addition to these effects, VPA reduced huntingtin aggregate deposition as an underlying cause of brain damage in HD, thereby reducing spontaneous locomotor activity. It is noteworthy that the administration of VPA in this study was performed before the onset of the disease to evaluate its preventive properties in HD, which may explain the conflicting results of this study with some other clinical studies in which VPA did not show efficacy in HD patients.90 In addition to this study, another investigation was conducted by Morland et al to assess the preventive effect of VPA in HD. In this study, which used malonate to induce HD, transgenic mice treated with VPA had a smaller brain lesion compared to other mice. In addition, it was shown that the rate of extracellular accumulation of glutamate in mice treated with VPA was lower compared to other mice.91 Due to the possible role of increased extracellular glutamate in the pathogenesis of HD disease, extracellular glutamate depletion was suggested as a neuroprotective mechanism of VPA, which may be due to increased glutamate transporter activity.92
One of the new therapeutic strategies for treating HD is stem cell therapy. A method used to increase the effectiveness of this treatment is the preconditioning of stem cells before transplantation.93 Therefore, Linares et al used VPA + Li for preconditioning of mesenchymal stem cells (MSCs) before transplanting to mice models of HD. This study showed that MSCs that were preconditioned with VPA + Li showed better migration and homing capacity and that more of these cells remained after transplantation and produced more neuroprotective factors (such as trophic and antiapoptotic factors), which generally improved motor function and survival in N171-82Q mice receiving this treatment. However, the use of this treatment did not improve the weight of the recipient mice, a failure that had been shown in previous studies using stem cells to treat HD mice.94 Another study that examined the efficacy of this combination therapy in HD transgenic mice was the study conducted by Chiu et al. In this study, the direct effect of VPA + Li on the improvement of symptoms in mice models of HD was evaluated. Two types of mice were used in this study including N171-82Q and 2AC128. It was shown that the use of this treatment led to improvement in movement disorder and depressive-like symptoms and an increase in motor skill learning and movement coordination. Interestingly, despite the ineffectiveness of Li or VPA monotherapy in reducing anxiety-like symptoms, treatment with both drugs reduced these symptoms. Among the neuroprotective factors that increased in the brain of studied mice were heat shock protein 70 (Hsp70)and BDNF. Contrary to the increase in the survival of mice under the influence of VPA + Li, this increase was not statistically significant, and as in the study of Linares, no favorable effects were seen on the weight of mice.95
Discussion
Based on the content provided, it can be concluded that besides the conventional use of VPA as an anticonvulsant drug, it can be used for other purposes including the treatment of patients with neurodegenerative disorders. However, the efficacy of this drug in different forms of neurodegenerative diseases is not the same and even different manifestations of these diseases do not respond equally to VPA. In addition, as mentioned earlier, the efficacy of VPA in neurodegenerative disease varies from study to study; therefore, definitive decisions about the use of this drug in patients with neurodegenerative disease need further investigations.
Conclusion
Besides, most of the studies in this field are only animal studies, the results of which cannot be directly generalized to the human population. However, according to many promising results in many studies, it seems that the use of this drug in the treatment of neurodegenerative patients may be beneficial. Figure 1 demonstrates the efficacy of VPA in neurodegenerative diseases. Table 1 summarizes the primary manifestation of neurodegenerative diseases and the effects of VPA on them.
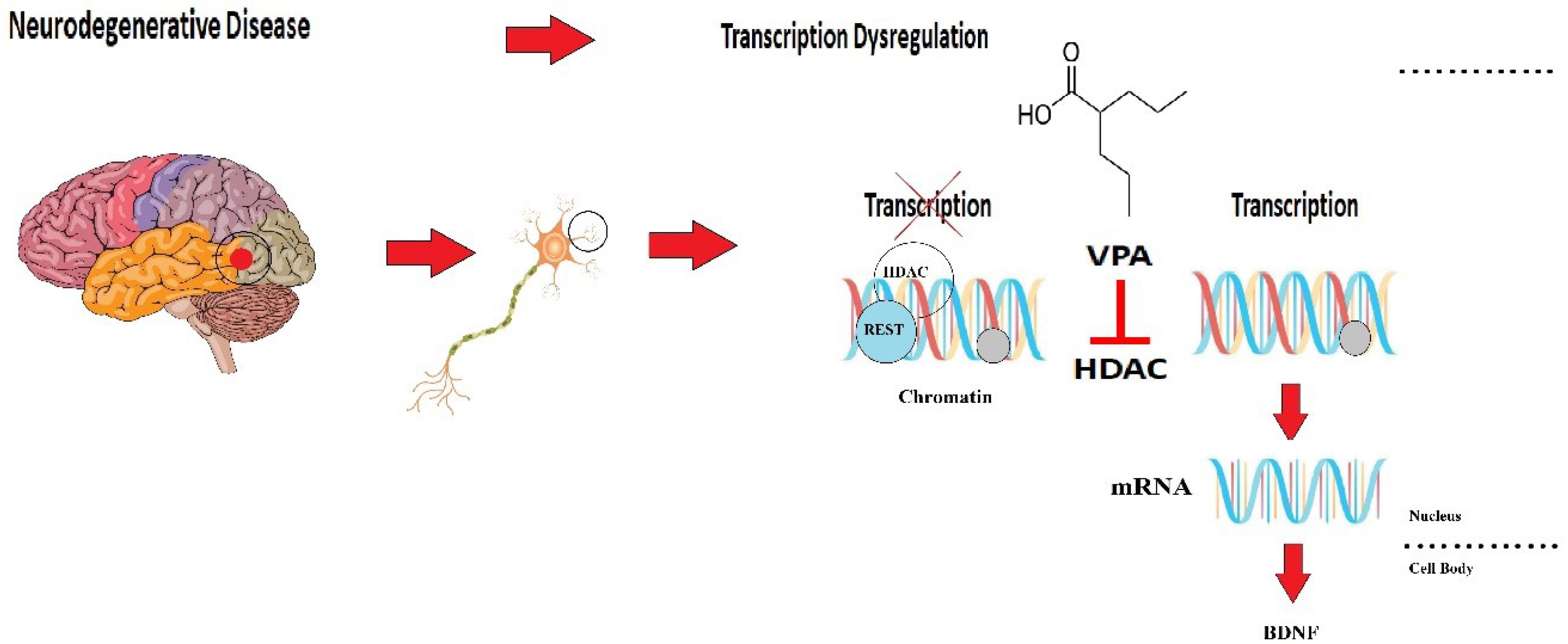
Figure 1.
Role of VPA in the Pathogenesis of Neurodegenerative Diseases by Affecting HDAC and BDNF
.
Role of VPA in the Pathogenesis of Neurodegenerative Diseases by Affecting HDAC and BDNF
Table 1.
Effects of Valproic Acid on Neurological Diseases
|
Disease
|
Characteristic of disease
|
Effect of valproic acid
|
| AD |
simple
-
-
✓ Production of inflammatory cytokines
-
✓ Phosphorylation of NFkB-p65 around the Aβ plaques
-
✓ Decreased melatonin production
-
✓ Tau protein dephosphorylation
-
✓ M2 (melatonin receptor) reduction
|
simple
-
✓ Increases anti-apoptotic factors including BCL-2 expression
-
✓ Reducing the production of inflammatory cytokines
-
✓ Prevents the formation of Aβ plaques
-
✓ Hyperphosphorylation of NFkB-p65 in the Aβ plaques
-
✓ Inhibits the GSK-3B activity through phosphorylation at serine-9 (producing phospho-GSK-3B-ser9)
-
✓ M2 (melatonin receptor) elevation
|
| Parkinson’s disease |
simple
-
✓ Destruction of dopaminergic cells
-
✓ Mutant alpha-synuclein (αSyn) formation
-
✓ Methyltransferase 1 increased in the cytoplasm and affected the promoter region of the αSyn gene (SNCA)
-
✓ Increased neuronal apoptosis through preventing histone acetylation
-
✓ Maintenance of tyrosine hydroxylase immunoreactivity in striatum and substantia nigra
|
simple
-
✓ Protects neurons against neurodegeneration
-
✓ Reverses the degeneration of dopaminergic nigrostriatal neurons
-
✓ Decreases midbrain volume and increases ventricular volume
-
-
-
✓ Increases BDNF, GDNF, and acetylation of histones
-
✓ Increases the levels of striatal dopamine
|
| MS |
simple
-
✓ Dysregulation of oxidative stress pathways, hormones, and excitotoxicity
-
✓ Dysregulated Th1 and Th17 cells
-
✓ ASK1 pathway dysregulation
-
✓ Unbalanced immune system affects astrocytes and causes demyelination
|
simple
-
✓ Prevents MS onset but increases the risk of developing MS in patients with a previous history of VPA use
-
✓ Reduces remyelination and oligodendrocyte precursors in the brain
-
✓ Reduces the infiltration of T cells into the optic nerve and spinal cord
-
✓ Suppresses the activity of glial cells, leading to T-cell suppression
|
| Huntington’s disease |
simple
-
✓ Replication of cytosine-adenine-guanine sequence in the Huntington (HTT, OMIM 613004) gene
-
-
✓ Decreased medium-sized GABAergic neurons in the striatum
-
✓ Increased histone deacetylation
-
✓ Increased extracellular glutamate
|
simple
-
✓ Enhances extracellular GABA
-
✓ Transgenic mice-treated VPA had a smaller brain lesion
-
✓ Lowering extracellular accumulation of glutamate
-
✓ Increases glutamate transporter activity
-
✓ Decreases histone deacetylation
-
✓ Increases survival rate in HD
|
Authors’ Contribution
Conceptualization: Hesameddin Razilou, Seyyed Sina Hejazian.
Data curation: Hesameddin Razilou, Seyyed Sina Hejazian.
Formal analysis: Hesameddin Razilou, Seyyed Sina Hejazian.
Investigation: Shaheh Meshkini.
Methodology: Shahed Meshkini.
Project administration: Hesameddin Razilou, Seyyed Sina Hejazian.
Resources: Hesameddin Razilou, Seyyed Sina Hejazian.
Software: Hesameddin Razilou, Seyyed Sina Hejazian.
Supervision: Shahed Meshkini.
Validation: Hesameddin Razilou, Seyyed Sina Hejazian.
Visualization: Hesameddin Razilou, Seyyed Sina Hejazian.
Writing–original draft: Hesameddin Razilou, Seyyed Sina Hejazian.
Writing–review & editing: Shahed Meshkini.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the current study.
Consent for Publication
Not applicable.
Ethical Approval
Not applicable.
References
- Rahman M, Nguyen H. Valproic acid. In: StatPearls [Internet]. Treasure Island, FL: StatPearls Publishing; 2020.
- Zhu MM, Li HL, Shi LH, Chen XP, Luo J, Zhang ZL. The pharmacogenomics of valproic acid. J Hum Genet 2017; 62(12):1009-14. doi: 10.1038/jhg.2017.91 [Crossref] [ Google Scholar]
- Mitra-Ghosh T, Callisto SP, Lamba JK, Remmel RP, Birnbaum AK, Barbarino JM. PharmGKB summary: lamotrigine pathway, pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics 2020; 30(4):81-90. doi: 10.1097/fpc.0000000000000397 [Crossref] [ Google Scholar]
- Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol 2010;2010. 10.1155/2010/479364.
- Jones GL, Popli GS, Silvia MT. Lacosamide-induced valproic acid toxicity. Pediatr Neurol 2013; 48(4):308-10. doi: 10.1016/j.pediatrneurol.2012.12.039 [Crossref] [ Google Scholar]
- Hussain R, Zubair H, Pursell S, Shahab M. Neurodegenerative diseases: regenerative mechanisms and novel therapeutic approaches. Brain Sci 2018; 8(9):177. doi: 10.3390/brainsci8090177 [Crossref] [ Google Scholar]
- Yu TW, Lane HY, Lin CH. Novel therapeutic approaches for Alzheimer’s disease: an updated review. Int J Mol Sci 2021; 22(15):8208. doi: 10.3390/ijms22158208 [Crossref] [ Google Scholar]
- Holman HR. The relation of the chronic disease epidemic to the health care crisis. ACR Open Rheumatol 2020; 2(3):167-73. doi: 10.1002/acr2.11114 [Crossref] [ Google Scholar]
- Stuckler D. Population causes and consequences of leading chronic diseases: a comparative analysis of prevailing explanations. Milbank Q 2008; 86(2):273-326. doi: 10.1111/j.1468-0009.2008.00522.x [Crossref] [ Google Scholar]
- Gilgun-Sherki Y, Melamed E, Offen D. Anti-inflammatory drugs in the treatment of neurodegenerative diseases: current state. Curr Pharm Des 2006; 12(27):3509-19. doi: 10.2174/138161206778343091 [Crossref] [ Google Scholar]
- Coppede F. Targeting the epigenome to treat neurodegenerative diseases or delay their onset: a perspective. Neural Regen Res 2022; 17(8):1745-7. doi: 10.4103/1673-5374.332145 [Crossref] [ Google Scholar]
- Chen S, Wu H, Klebe D, Hong Y, Zhang J. Valproic acid: a new candidate of therapeutic application for the acute central nervous system injuries. Neurochem Res 2014; 39(9):1621-33. doi: 10.1007/s11064-014-1241-2 [Crossref] [ Google Scholar]
- Dong D, Xie J, Wang J. Neuroprotective effects of brain-gut peptides: a potential therapy for Parkinson’s disease. Neurosci Bull 2019; 35(6):1085-96. doi: 10.1007/s12264-019-00407-3 [Crossref] [ Google Scholar]
- Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci 2006; 26(28):7502-12. doi: 10.1523/jneurosci.0096-06.2006 [Crossref] [ Google Scholar]
- Zhang C, Yuan X, Hu Z, Liu S, Li H, Wu M. Valproic acid protects primary dopamine neurons from MPP( + )-induced neurotoxicity: involvement of GSK3β phosphorylation by Akt and ERK through the mitochondrial intrinsic apoptotic pathway. Biomed Res Int 2017; 2017:8124501. doi: 10.1155/2017/8124501 [Crossref] [ Google Scholar]
- Leng Y, Chuang DM. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci 2006; 26(28):7502-12. doi: 10.1523/jneurosci.0096-06.2006 [Crossref] [ Google Scholar]
- Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C. Inhibition of GSK-3 ameliorates Aβ pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet 2010; 6(9):e1001087. doi: 10.1371/journal.pgen.1001087 [Crossref] [ Google Scholar]
- Jayaraman S, Jayaraman A. Impact of histone modifier-induced protection against autoimmune encephalomyelitis on multiple sclerosis treatment. Front Neurol 2022; 13:980758. doi: 10.3389/fneur.2022.980758 [Crossref] [ Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26(40):10129-40. doi: 10.1523/jneurosci.1202-06.2006 [Crossref] [ Google Scholar]
- Lv J, Du C, Wei W, Wu Z, Zhao G, Li Z. The antiepileptic drug valproic acid restores T cell homeostasis and ameliorates pathogenesis of experimental autoimmune encephalomyelitis. J Biol Chem 2012; 287(34):28656-65. doi: 10.1074/jbc.M112.356584 [Crossref] [ Google Scholar]
- Zhang Y, Yin HY, Rubini P, Tang Y, Illes P. A Possible Causal Involvement of Neuroinflammatory, Purinergic P2X7 Receptors in Psychiatric Disorders. Curr Neuropharmacol 2022; 20(11):2142-55. doi: 10.2174/1570159x20666220302152400S [Crossref] [ Google Scholar]
- ingh D, Gupta S, Verma I, Morsy MA, Nair AB, Ahmed AF. Hidden pharmacological activities of valproic acid: a new insight. Biomed Pharmacother 2021; 142:112021. doi: 10.1016/j.biopha.2021.112021 [Crossref] [ Google Scholar]
- Breijyeh Z, Karaman R. Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 2020; 25(24):5789. doi: 10.3390/molecules25245789 [Crossref] [ Google Scholar]
- Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J Alzheimers Dis 2017; 57(4):975-99. doi: 10.3233/jad-160612 [Crossref] [ Google Scholar]
- Lebois EP, Thorn C, Edgerton JR, Popiolek M, Xi S. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018; 136(Pt C):362-73. doi: 10.1016/j.neuropharm.2017.11.018 [Crossref] [ Google Scholar]
- Javaid SF, Giebel C, Khan MA, Hashim MJ. Epidemiology of Alzheimer’s disease and other dementias: rising global burden and forecasted trends. F1000Res 2021; 10:425. doi: 10.12688/f1000research.50786.1 [Crossref] [ Google Scholar]
- Torrisi M, Cacciola A, Marra A, De Luca R, Bramanti P, Calabrò RS. Inappropriate behaviors and hypersexuality in individuals with dementia: an overview of a neglected issue. Geriatr Gerontol Int 2017; 17(6):865-74. doi: 10.1111/ggi.12854 [Crossref] [ Google Scholar]
- Ohno Y, Kunisawa N, Shimizu S. Antipsychotic treatment of behavioral and psychological symptoms of dementia (BPSD): management of extrapyramidal side effects. Front Pharmacol 2019; 10:1045. doi: 10.3389/fphar.2019.01045 [Crossref] [ Google Scholar]
- Sgobio C, Ghiglieri V, Costa C, Bagetta V, Siliquini S, Barone I. Hippocampal synaptic plasticity, memory, and epilepsy: effects of long-term valproic acid treatment. Biol Psychiatry 2010; 67(6):567-74. doi: 10.1016/j.biopsych.2009.11.008 [Crossref] [ Google Scholar]
- Yao ZG, Liang L, Liu Y, Zhang L, Zhu H, Huang L. Valproate improves memory deficits in an Alzheimer’s disease mouse model: investigation of possible mechanisms of action. Cell Mol Neurobiol 2014; 34(6):805-12. doi: 10.1007/s10571-013-0012-y [Crossref] [ Google Scholar]
- Wang Z, Yang D, Zhang X, Li T, Li J, Tang Y. Hypoxia-induced down-regulation of neprilysin by histone modification in mouse primary cortical and hippocampal neurons. PLoS One 2011; 6(4):e19229. doi: 10.1371/journal.pone.0019229 [Crossref] [ Google Scholar]
- Tan C, Xuan L, Cao S, Yu G, Hou Q, Wang H. Decreased histone deacetylase 2 (HDAC2) in peripheral blood monocytes (PBMCs) of COPD patients. PLoS One 2016; 11(1):e0147380. doi: 10.1371/journal.pone.0147380 [Crossref] [ Google Scholar]
- Nalivaeva NN, Belyaev ND, Lewis DI, Pickles AR, Makova NZ, Bagrova DI. Effect of sodium valproate administration on brain neprilysin expression and memory in rats. J Mol Neurosci 2012; 46(3):569-77. doi: 10.1007/s12031-011-9644-x [Crossref] [ Google Scholar]
- Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F. Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med 2008; 205(12):2781-9. doi: 10.1084/jem.20081588 [Crossref] [ Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 1987; 79(1-2):195-200. doi: 10.1016/0304-3940(87)90696-3 [Crossref] [ Google Scholar]
- Kosugi K, Yoshida K, Suzuki T, Kobayashi K, Yoshida K, Mimura M. Activation of ventral CA1 hippocampal neurons projecting to the lateral septum during feeding. Hippocampus 2021; 31(3):294-304. doi: 10.1002/hipo.23289 [Crossref] [ Google Scholar]
- Sans-Dublanc A, Razzauti A, Desikan S, Pascual M, Monyer H, Sindreu C. Septal GABAergic inputs to CA1 govern contextual memory retrieval. Sci Adv 2020; 6(44):eaba5003. doi: 10.1126/sciadv.aba5003 [Crossref] [ Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer’s disease. CNS Neurol Disord Drug Targets 2010; 9(2):156-67. doi: 10.2174/187152710791012071 [Crossref] [ Google Scholar]
- Xuan A, Long D, Li J, Ji W, Hong L, Zhang M. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci 2012; 90(11-12):463-8. doi: 10.1016/j.lfs.2012.01.001 [Crossref] [ Google Scholar]
- Cavallucci V, D’Amelio M, Cecconi F. Aβ toxicity in Alzheimer’s disease. Mol Neurobiol 2012; 45(2):366-78. doi: 10.1007/s12035-012-8251-3 [Crossref] [ Google Scholar]
- Zhang F, Jiang L. Neuroinflammation in Alzheimer’s disease. Neuropsychiatr Dis Treat 2015; 11:243-56. doi: 10.2147/ndt.S75546 [Crossref] [ Google Scholar]
- Xuan AG, Pan XB, Wei P, Ji WD, Zhang WJ, Liu JH. Valproic acid alleviates memory deficits and attenuates amyloid-β deposition in transgenic mouse model of Alzheimer’s disease. Mol Neurobiol 2015; 51(1):300-12. doi: 10.1007/s12035-014-8751-4 [Crossref] [ Google Scholar]
- Kirouac L, Rajic AJ, Cribbs DH, Padmanabhan J. Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro 2017;4(2):ENEURO.0149-16.2017. 10.1523/eneuro.0149-16.2017.
- Christian F, Smith EL, Carmody RJ. The regulation of NF-κB subunits by phosphorylation. Cells 2016; 5(1):12. doi: 10.3390/cells5010012 [Crossref] [ Google Scholar]
- Chen S, Ye J, Chen X, Shi J, Wu W, Lin W. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-κB pathway dependent of HDAC3. J Neuroinflammation 2018; 15(1):150. doi: 10.1186/s12974-018-1193-6 [Crossref] [ Google Scholar]
- Long Z, Zeng Q, Wang K, Sharma A, He G. Gender difference in valproic acid-induced neuroprotective effects on APP/PS1 double transgenic mice modeling Alzheimer’s disease. Acta Biochim Biophys Sin (Shanghai) 2016; 48(10):930-8. doi: 10.1093/abbs/gmw085 [Crossref] [ Google Scholar]
- Ornoy A, Weinstein-Fudim L, Ergaz Z. Prevention or amelioration of autism-like symptoms in animal models: will it bring us closer to treating human ASD?. Int J Mol Sci 2019; 20(5):1074. doi: 10.3390/ijms20051074 [Crossref] [ Google Scholar]
- Shukla M, Govitrapong P, Boontem P, Reiter RJ, Satayavivad J. Mechanisms of melatonin in alleviating Alzheimer’s disease. Curr Neuropharmacol 2017; 15(7):1010-31. doi: 10.2174/1570159x15666170313123454 [Crossref] [ Google Scholar]
- Song J. Pineal gland dysfunction in Alzheimer’s disease: relationship with the immune-pineal axis, sleep disturbance, and neurogenesis. Mol Neurodegener 2019; 14(1):28. doi: 10.1186/s13024-019-0330-8 [Crossref] [ Google Scholar]
- Yang X, Chen Q. Efficacy of the combined use of donepezil with either quetiapine or sodium valproate in patients with Alzheimer’s disease with behavioral and psychological symptoms of dementia, and their effects on vascular endothelial growth factors. Exp Ther Med 2021; 21(1):10. doi: 10.3892/etm.2020.9442 [Crossref] [ Google Scholar]
- Herrmann N, Lanctôt KL, Rothenburg LS, Eryavec G. A placebo-controlled trial of valproate for agitation and aggression in Alzheimer’s disease. Dement Geriatr Cogn Disord 2007; 23(2):116-9. doi: 10.1159/000097757 [Crossref] [ Google Scholar]
- Hirschfeld RM, Bowden CL, Vigna NV, Wozniak P, Collins M. A randomized, placebo-controlled, multicenter study of divalproex sodium extended-release in the acute treatment of mania. J Clin Psychiatry 2010; 71(4):426-32. doi: 10.4088/JCP.08m04960yel [Crossref] [ Google Scholar]
- Kouli A, Torsney KM, Kuan WL. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. Exon Publications; 2018. p. 3-26.
- Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet 2021; 397(10291):2284-303. doi: 10.1016/s0140-6736(21)00218-x [Crossref] [ Google Scholar]
- Sveinbjornsdottir S. The clinical symptoms of Parkinson’s disease. J Neurochem 2016; 139 Suppl 1:318-24. doi: 10.1111/jnc.13691 [Crossref] [ Google Scholar]
- Labbé C, Lorenzo-Betancor O, Ross OA. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol 2016; 132(4):515-30. doi: 10.1007/s00401-016-1590-9 [Crossref] [ Google Scholar]
- Riederer P, Berg D, Casadei N, Cheng F, Classen J, Dresel C. α-Synuclein in Parkinson’s disease: causal or bystander?. J Neural Transm 2019; 126(7):815-40. doi: 10.1007/s00702-019-02025-9 [Crossref] [ Google Scholar]
- Karimi-Moghadam A, Charsouei S, Bell B, Jabalameli MR. Parkinson disease from mendelian forms to genetic susceptibility: new molecular insights into the neurodegeneration process. Cell Mol Neurobiol 2018; 38(6):1153-78. doi: 10.1007/s10571-018-0587-4 [Crossref] [ Google Scholar]
- Averbeck BB, O’Sullivan SS, Djamshidian A. Impulsive and compulsive behaviors in Parkinson’s disease. Annu Rev Clin Psychol 2014; 10:553-80. doi: 10.1146/annurev-clinpsy-032813-153705 [Crossref] [ Google Scholar]
- Gatto EM, Aldinio V. Impulse control disorders in Parkinson’s disease A brief and comprehensive review. Front Neurol 2019; 10:351. doi: 10.3389/fneur.2019.00351 [Crossref] [ Google Scholar]
- Watanabe S, Hori T, Karita S, Hayashi T, Nishitoba J, Unno Y, Soplin NE, Heymann J, Wiesner M, Chen N, Renduchintala A. ESPnet: End-to-end speech processing toolkit. Preprint. Posted online March 30, 2018. arXiv:1804.00015. 10.48550/arXiv.1804.00015.
- Lai CL, Lu CC, Lin HC, Sung YF, Wu YP, Hong JS. Valproate is protective against 6-OHDA-induced dopaminergic neurodegeneration in rodent midbrain: a potential role of BDNF up-regulation. J Formos Med Assoc 2019; 118(1 Pt 3):420-8. doi: 10.1016/j.jfma.2018.06.017 [Crossref] [ Google Scholar]
- Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2011; 194:189-94. doi: 10.1016/j.neuroscience.2011.08.010 [Crossref] [ Google Scholar]
- Drui G, Carnicella S, Carcenac C, Favier M, Bertrand A, Boulet S. Loss of dopaminergic nigrostriatal neurons accounts for the motivational and affective deficits in Parkinson’s disease. Mol Psychiatry 2014; 19(3):358-67. doi: 10.1038/mp.2013.3 [Crossref] [ Google Scholar]
- Harrison IF, Crum WR, Vernon AC, Dexter DT. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br J Pharmacol 2015; 172(16):4200-15. doi: 10.1111/bph.13208 [Crossref] [ Google Scholar]
- Kim T, Song S, Park Y, Kang S, Seo H. HDAC inhibition by valproic acid induces neuroprotection and improvement of PD-like behaviors in LRRK2 R1441G transgenic mice. Exp Neurobiol 2019; 28(4):504-15. doi: 10.5607/en.2019.28.4.504 [Crossref] [ Google Scholar]
- Buzgoova K, Graban J, Balagova L, Hlavacova N, Jezova D. Brain derived neurotrophic factor expression and DNA methylation in response to subchronic valproic acid and/or aldosterone treatment. Croat Med J 2019; 60(2):71-7. doi: 10.3325/cmj.2019.60.71 [Crossref] [ Google Scholar]
- Carriere CH, Kang NH, Niles LP. Neuroprotection by valproic acid in an intrastriatal rotenone model of Parkinson’s disease. Neuroscience 2014; 267:114-21. doi: 10.1016/j.neuroscience.2014.02.028 [Crossref] [ Google Scholar]
- Brichta L, Greengard P, Flajolet M. Advances in the pharmacological treatment of Parkinson’s disease: targeting neurotransmitter systems Trends in neurosciences. 2013 S ep 1; 36(9):543-54. [ Google Scholar]
- Castro AA, Ghisoni K, Latini A, Quevedo J, Tasca CI, Prediger RD. Lithium and valproate prevent olfactory discrimination and short-term memory impairments in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) rat model of Parkinson’s disease. Behav Brain Res 2012; 229(1):208-15. doi: 10.1016/j.bbr.2012.01.016 [Crossref] [ Google Scholar]
- Roostalu U, Salinas CBG, Thorbek DD, Skytte JL, Fabricius K, Barkholt P. Quantitative whole-brain 3D imaging of tyrosine hydroxylase-labeled neuron architecture in the mouse MPTP model of Parkinson’s disease. Dis Model Mech 2019; 12(11):dmm042200. doi: 10.1242/dmm.042200 [Crossref] [ Google Scholar]
- Degirmenci Y, Kececi H. Pathological gambling in a patient with Parkinson’s disease and valproate response. Dusunen Adam 2019; 32(4):365-7. doi: 10.14744/dajpns.2019.00053 [Crossref] [ Google Scholar]
- O’Sullivan SS, Evans AH, Lees AJ. Dopamine dysregulation syndrome: an overview of its epidemiology, mechanisms and management. CNS Drugs 2009; 23(2):157-70. doi: 10.2165/00023210-200923020-00005 [Crossref] [ Google Scholar]
- Barbosa P, Djamshidian A, Lees AJ, Warner TT. The outcome of dopamine dysregulation syndrome in Parkinson’s disease: a retrospective postmortem study. Mov Disord Clin Pract 2018; 5(5):519-22. doi: 10.1002/mdc3.12671 [Crossref] [ Google Scholar]
- Sriram A, Ward HE, Hassan A, Iyer S, Foote KD, Rodriguez RL. Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson’s disease. J Neurol 2013; 260(2):521-7. doi: 10.1007/s00415-012-6669-1 [Crossref] [ Google Scholar]
- Hicks CW, Pandya MM, Itin I, Fernandez HH. Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease. Parkinsonism Relat Disord 2011; 17(5):379-81. doi: 10.1016/j.parkreldis.2011.03.003 [Crossref] [ Google Scholar]
- Grall-Bronnec M, Victorri-Vigneau C, Donnio Y, Leboucher J, Rousselet M, Thiabaud E. Dopamine agonists and impulse control disorders: a complex association. Drug Saf 2018; 41(1):19-75. doi: 10.1007/s40264-017-0590-6 [Crossref] [ Google Scholar]
- Romoli M, Mazzocchetti P, D’Alonzo R, Siliquini S, Rinaldi VE, Verrotti A. Valproic acid and epilepsy: from molecular mechanisms to clinical evidences. Curr Neuropharmacol 2019; 17(10):926-46. doi: 10.2174/1570159x17666181227165722 [Crossref] [ Google Scholar]
- Ximenes JC, Neves KR, Leal LK, do Carmo MR, de Castro Brito GA, da Graça Naffah-Mazzacoratti M. Valproic acid neuroprotection in the 6-OHDA model of Parkinson’s disease is possibly related to its anti-inflammatory and HDAC inhibitory properties. J Neurodegener Dis 2015; 2015:313702. doi: 10.1155/2015/313702 [Crossref] [ Google Scholar]
- Muralidharan A, Rahman J, Banerjee D, Hakim Mohammed AR, Malik BH. Parkinsonism: a rare adverse effect of valproic acid. Cureus 2020; 12(6):e8782. doi: 10.7759/cureus.8782 [Crossref] [ Google Scholar]
- Salazar Z, Tschopp L, Calandra C, Micheli F. Pisa syndrome and parkinsonism secondary to valproic acid in Huntington’s disease. Mov Disord 2008; 23(16):2430-1. doi: 10.1002/mds.22328 [Crossref] [ Google Scholar]
- Dobson R, Giovannoni G. Multiple sclerosis - a review. Eur J Neurol 2019; 26(1):27-40. doi: 10.1111/ene.13819 [Crossref] [ Google Scholar]
- Amini ME, Shomali N, Bakhshi A, Rezaei S, Hemmatzadeh M, Hosseinzadeh R. Gut microbiome and multiple sclerosis: new insights and perspective. Int Immunopharmacol 2020; 88:107024. doi: 10.1016/j.intimp.2020.107024 [Crossref] [ Google Scholar]
- Buscarinu MC, Reniè R, Morena E, Romano C, Bellucci G, Marrone A. Late-onset MS: disease course and safety-efficacy of DMTS. Front Neurol 2022; 13:829331. doi: 10.3389/fneur.2022.829331 [Crossref] [ Google Scholar]
- Ghaderian S, Shomali N, Behravesh S, Rezaei Danbaran G, Hemmatzadeh M, Aslani S. The emerging role of lncRNAs in multiple sclerosis. J Neuroimmunol 2020; 347:577347. doi: 10.1016/j.jneuroim.2020.577347 [Crossref] [ Google Scholar]
- Manouchehri N, Zhang Y, Salter A, Hussain RZ, Hartung HP, Hemmer B. Clinical trials in multiple sclerosis: potential future trial designs. Ther Adv Neurol Disord 2019; 12:1756286419847095. doi: 10.1177/1756286419847095 [Crossref] [ Google Scholar]
- Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis - success from bench to bedside. Nat Rev Neurol 2019; 15(1):53-8. doi: 10.1038/s41582-018-0082-z [Crossref] [ Google Scholar]
- Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017; 140(3):527-46. doi: 10.1093/brain/aww258 [Crossref] [ Google Scholar]
- Shirani A, Okuda DT, Stüve O. Therapeutic advances and future prospects in progressive forms of multiple sclerosis. Neurotherapeutics 2016; 13(1):58-69. doi: 10.1007/s13311-015-0409-z [Crossref] [ Google Scholar]
- Mulero P, Midaglia L, Montalban X. Ocrelizumab: a new milestone in multiple sclerosis therapy. Ther Adv Neurol Disord 2018; 11:1756286418773025. doi: 10.1177/1756286418773025 [Crossref] [ Google Scholar]
- Zhang Y, Salter A, Wallström E, Cutter G, Stüve O. Evolution of clinical trials in multiple sclerosis. Ther Adv Neurol Disord 2019; 12:1756286419826547. doi: 10.1177/1756286419826547 [Crossref] [ Google Scholar]
- Rossi M, Petralla S, Protti M, Baiula M, Kobrlova T, Soukup O. α-Linolenic acid-valproic acid conjugates: toward single-molecule polypharmacology for multiple sclerosis. ACS Med Chem Lett 2020; 11(12):2406-13. doi: 10.1021/acsmedchemlett.0c00375 [Crossref] [ Google Scholar]
- Yang L, Tsai TH. Neuroprotective effect and herbal-drug pharmacokinetic interaction of Gastrodiaelata extract on valproic acid. Biomed Pharmacother 2022; 156:113938. doi: 10.1016/j.biopha.2022.113938 [Crossref] [ Google Scholar]
- Nielsen NM, Svanström H, Stenager E, Magyari M, Koch-Henriksen N, Pasternak B. The use of valproic acid and multiple sclerosis. Pharmacoepidemiol Drug Saf 2015; 24(3):262-8. doi: 10.1002/pds.3692 [Crossref] [ Google Scholar]
- Johnson B, Nichols S. Crying and suicidal, but not depressed Pseudobulbar affect in multiple sclerosis successfully treated with valproic acid: case report and literature review. Palliat Support Care 2015; 13(6):1797-801. doi: 10.1017/s1478951514000376 [Crossref] [ Google Scholar]