Biomed Res Bull. 1(1):30-37.
doi: 10.34172/biomedrb.2023.07
Review Article
A Comprehensive and Updated Review of MicroRNA Pathogenesis and Gene Expression in Cystic Fibrosis
Delaram Mehrzadeh 1  , Mahsa Shorodi 1, Sina Rahimpour 1, *
, Mahsa Shorodi 1, Sina Rahimpour 1, *
Author information:
1Immunology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Abstract
MicroRNAs (miRNAs) are short (~22 nucleotides) non-coding sequences that regulate gene expression. It has been recently identified that miR-145 regulates cystic fibrosis transmembrane regulator (CFTR) expression, and response to first-generation miRNAs is recognized as major biomedical research in CF. Abnormal miRNA activity causes numerous diseases, thus the critical role of miRNA is to focus on miRNA bindings to target proteins. miRNA classifications depend on their genomic location and gene structure, and they include intergenic and intronic regions. Intronic miRNA location is in the introns of annotated genes. Intergenic and intronic miRNA genes are controlled with their promoters. Some miRNAs negatively regulate the cystic fibrosis transmembrane conductance regulator (CFTR) gene expression (e.g., miR-101) in adult lung cells and have no effect on fetal epithelial cells. MiR-145, miR-223, and miR-494 directly regulate the CFTR gene expression. MiR-509-3p and miR-494 regulate CFTR more strongly than the other miRNAs. MiR-138 enhances CFTR channels on the cell surface of epithelial cells. Polymorphisms in the 3′-UTR region of CFTR decrease the expression of CFTR. Mutations in the CF (CFTR) gene, located on chromosome 7, diminish ion transport. The consequent loss of chloride and bicarbonate secretion and absorption lead to a multi-organ disease called CF marked by mucus inspissation, chronic inflammation, and tissue destruction. The most affected organs are the lung and pancreas, but the liver, bowel, sinuses, and vas deferens, among others, are prominently involved as well. The loss of CFTR also results in an altered miRNA profile that regulates the response to therapy and disease pathogenesis. This review summarizes miRNA alterations in the CF tissue, the role of miRNA in disease pathogenesis, and the therapeutic opportunity for oligotherapeutic manipulation to improve CF outcomes.
Keywords: MicroRNA, Cystic fibrosis, Gene expression
Copyright and License Information
© 2023 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Introduction
miRNA properties, biogenesis, and function
microRNAs (miRNAs) are small non-coding RNAs that indicate a primary post-transcriptional gene regulation mechanism and modulate all biological processes.1 miRNAs suppress gene expression by binding to the 3′ untranslated region (UTR) of a messenger RNA (mRNA). The profiles of miRNA expression are changed between health and disease via altered biomarkers of disease. Studies suggested that miRNAs are potentially crucial in most pulmonary diseases such as asthma, chronic obstructive pulmonary disease, and cystic fibrosis (CF).2,3 The biogenesis of miRNAs starts from the nucleus, finishes in the cytoplasm, and involves several enzymes and proteins.4 In the core, the primary miRNA transcript (pri-miRNA) is modified with a 5’ 7-methylguanosine cap and a 3’ poly-A tail and transcribed by RNA polymerase II or III.5 The microprocessor complex, consisting of double-stranded RNA-binding protein, DGCR8, and Drosha, recognizes pri-miRNAs in the nucleus and excises it to yield a 70 nucleotide that is transported from the nucleus to cytoplasm through Exportin 5 and a RAS-related nuclear protein.6 In the cytoplasm, the pre-miRNA is processed by Dicer, a second RNase III enzyme, to ~22-nucleotide (nt) miRNA duplex containing the mature miRNA. Argonaut proteins recognize the duplex and then combinate the mature miRNA to the RNA-induced silencing complex (RISC).7 The RISC complex transports miRNA to the 3′ UTR of a messenger RNA (mRNA). MiRNAs bind to AU-rich elements (AREs) and block ARE-binding proteins to alter gene expression.8 Mature miRNAs use the 3’UTR to selectively bind to miRNA response elements (MRE). The MRE motif can be found on multiple different mRNAs. The repression of transcripts occurs at the 5’UTR region by using a luciferase reporter including diverse wild-type and 5′UTRs mutants from CFTR mRNA transcripts, which are 132nt 5′UTR (CFTR-132) in fetal lung, to 5′UTR, which is -69nt (CFTR-69) in adults. This biogenesis causes miRNAs to become a new diagnostic, therapeutic tool for the early detection, classification, and treatment of human diseases.9,10
The primary function of miRNAs in gene therapy affect target genes and modulates diverse signaling pathways. The regulation of gene expression occurs with miRNAs at transcription and translation processes.10 The first stage is also known as the pretranslation step which includes chromatin remodeling. miRNA interplay with the promoter region can induce transcription. miRNAs identify mRNAs targets for repression by the localized multi-protein nucleus which is called miRISC. Nucleus miRISC regulates the transcriptional levels of mRNA and activates transcription. In addition, these proteins can directly regulate transcription. miRISC promotes chromatin states and splicing profiles by DNA and mRNA interaction. Cytoplasmic miRISC can diffuse to the cytosol and promote translation activation.
The post-initiation step is the second stage in which miRNAs modulate gene expression. The hypermethylation of miRNA genes as a transcriptional regulation and miRNA processing and stability changing as a post-transcriptional are important in miRNA expression caused by endogenous and exogenous compounds. miRNA expression downregulates at the post-transcriptional level due to miRNA biogenesis enzymes affected by mutations. Further, mRNA degradation and translational repression occur at this level. Moreover, miRNAs suppress the expression of target genes by interacting with the 3’UTR.
Furthermore, ribosomal subunit dissociation and premature termination occur in this step. The primary mechanism of this stage is the hybridization of miRNA-mRNA, which causes gene regulation. miRNA interaction with mRNA is due to Watson-Crick pairing.11
Cystic fibrosis
Structure and mechanical functioning of CFTR
The protein sequence of the CFTR is a combination of 1480 amino acids. It starts from two repeated motifs that implicate a hydrophilic membrane-spanning domain (MSD), including six helices and the central hydrophilic region for binding with adenosine triphosphate(ATP). These motifs are bound together with cytoplasmic (the R domain), which is encoded by exon 13. The primary structure of the CFTR protein looks like the members of membrane protein families such as ABC transporters (ATP-binding cassettes), which are the transport activators of substrates across the cell membrane. The function of CFTR revolves around two possibilities; the CFTR protein is a Cl-channel, and the other is the role of CFTR in the regulation of the Cl-channel (Figure 1).12 The CFTR structure includes five domains, including two MSDs (MSD-1 and −2), two nucleotide-binding domains (NBD-1 and -2), and a regulatory domain (the R domain). Each of the MSD-1 and -2 consists of six transmembrane helices include TM1-6 for MSD-1 and TM7-12 for MSD-2, respectively. The unique structure of the CFTR gene is the connection of MSD-2 and NBD-1 modules to the R domain, making a regulatory region with disorders that cannot be found in the other transporters. R-domain phosphorylation controls the activity of the chloride channel. The R area consists of numerous phosphorylation sites for protein kinase A (PKA), a cyclic AMP-dependent kinase that is notably conserved among species. PKA is activated when the intracellular concentration of chloride ions increases, opening the channel. Similarly, extracellular chloride absorption regulates channel gating, and increased extracellular chloride concentrations promote channel opening. Therefore, the CFTR facilitates the bidirectional permeability of chloride ions. The carboxyl-terminal, including threonine, arginine, and leucine is attached via a PDZ-like binding interplay with the cytoskeleton. The NBDs, R domain, and the N- and C- termini are intracellular. The CFTR transporter features is a regulated adenosine 3’,5’-monophosphate (cAMP) chloride channel in exocrine epithelia.
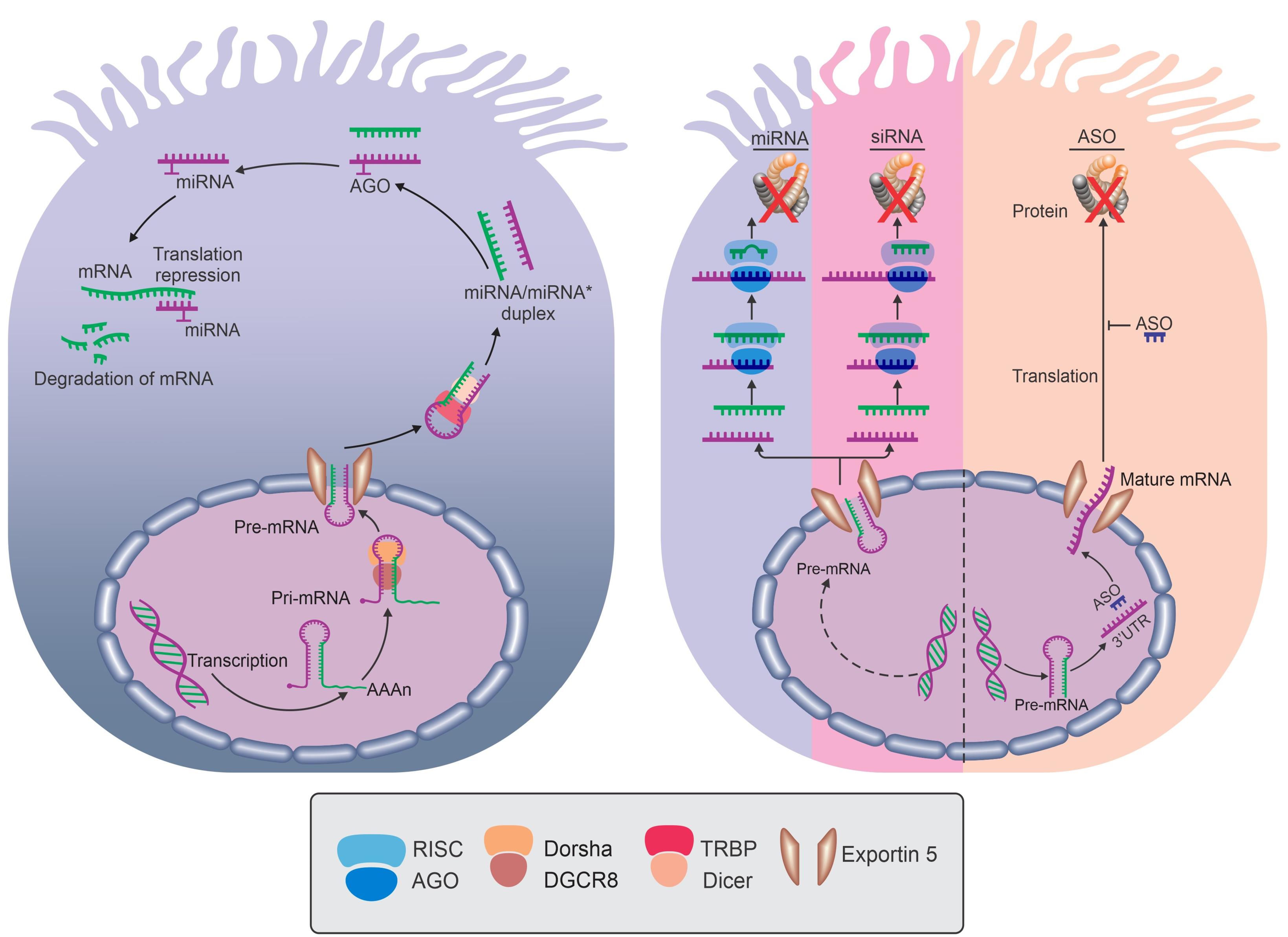
Figure 1.
Structure and Function of CFTR. Note. CFTR: Cystic fibrosis transmembrane regulator; AGO: Argonaut; ASO: Antisense oligonucleotide; miRNA: microRNA; mRNA: Messenger RNA; siRNA: Short interfering RNA; Pre-mRNA: Precursor messenger RNA; RISC: RNA-induced silencing complex; TRBP: TAR RNA binding protein; DGCR8: DiGeorge syndrome critical region 8.
.
Structure and Function of CFTR. Note. CFTR: Cystic fibrosis transmembrane regulator; AGO: Argonaut; ASO: Antisense oligonucleotide; miRNA: microRNA; mRNA: Messenger RNA; siRNA: Short interfering RNA; Pre-mRNA: Precursor messenger RNA; RISC: RNA-induced silencing complex; TRBP: TAR RNA binding protein; DGCR8: DiGeorge syndrome critical region 8.
Based on further studies, the first postulated is true. Additionally, CFTR proteins regulate the outwardly rectifying chloride channel, epithelial Na + channel (ENaC), and K + channels (Figure 2A). The primary function of the CFTR protein is ATP transportation and the regulation of the pH of intracellular organelles. The mutations of CFTR genes make anomalies in the effects of the CFTR protein which is related to the Cl-channel function. Welsh and Smith classified CFTR gene mutations into six classes.
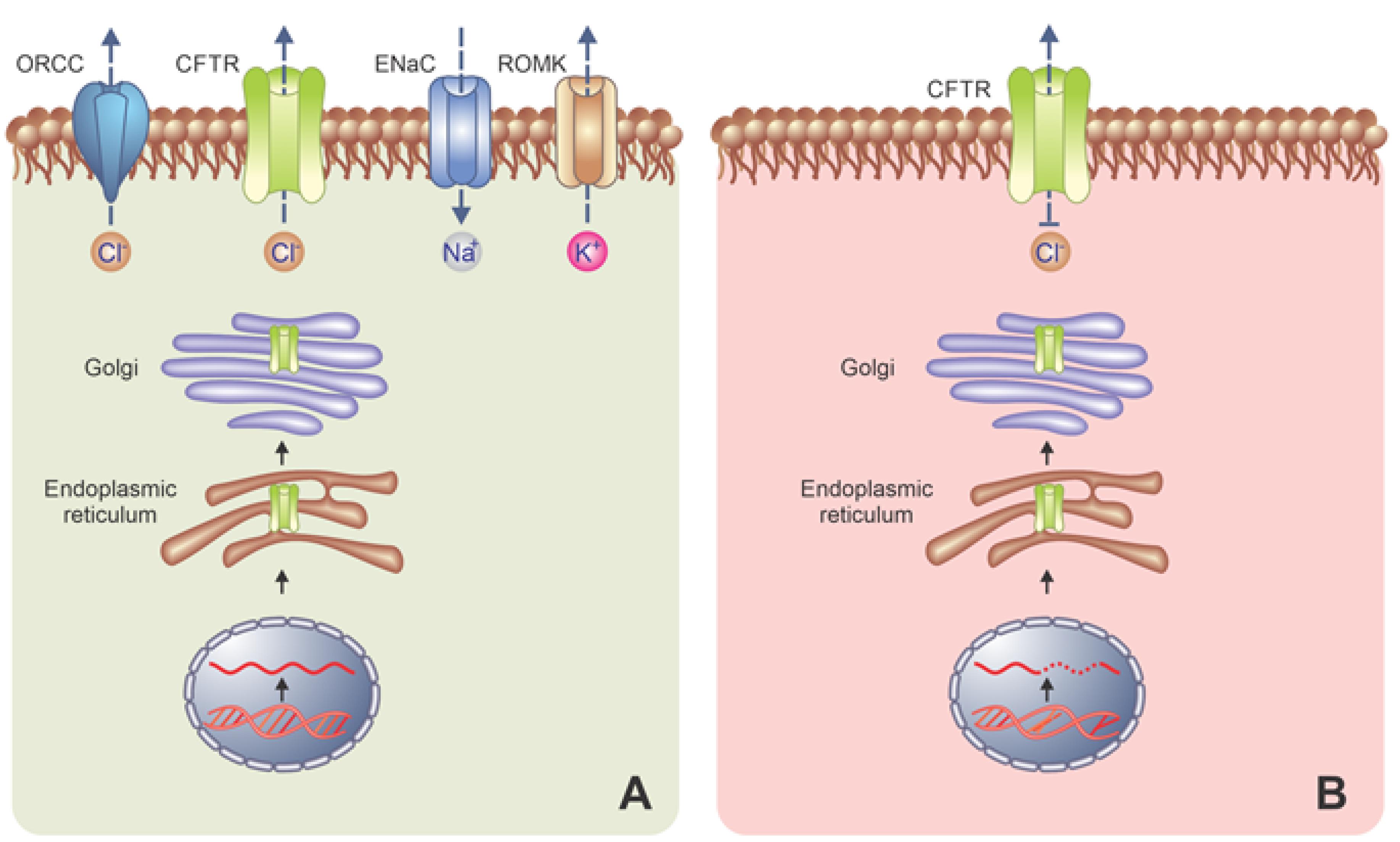
Figure 2.
(A) CFTR proteins regulate the epithelial Na+ channel and K+ channels and (B) class i- mutations altering production, class II- mutations altering maturation, class III- altering regulation, class IV-altering Conduction, Class V-mut altering the Stability of mRNA, class VI- mutations altering the stability of mature CFTR protein. Note. ENaC: Epithelial sodium channel; CFTR: Cystic fibrosis transmembrane regulator; ORCC: Outwardly rectifying chloride channel. ROMK: Renal outer medullary potassium; mRNA: Messenger RNA.
.
(A) CFTR proteins regulate the epithelial Na+ channel and K+ channels and (B) class i- mutations altering production, class II- mutations altering maturation, class III- altering regulation, class IV-altering Conduction, Class V-mut altering the Stability of mRNA, class VI- mutations altering the stability of mature CFTR protein. Note. ENaC: Epithelial sodium channel; CFTR: Cystic fibrosis transmembrane regulator; ORCC: Outwardly rectifying chloride channel. ROMK: Renal outer medullary potassium; mRNA: Messenger RNA.
Type I mutation is associated with protein production. Generally, the protein is not produced at all or in minimal quantities. These mutations consist of transcript processing. Class II mutations prevent the maturation of protein which undergoes degradation before being translocated to the apical membrane. In class III and IV mutations, protein is produced but works incorrectly. In class III, unusual ATP binding and hydrolysis occur in chloride channels. Class V mutations allow for the slow production of small quantities of a functional protein (mutations altering the stability of mRNA), thus class V mutations are associated with the reduced number of CFTR mRNA, and class VI are mutations altering the strength of mature CFTR protein (Figure 2B).
Overall, class I includes nonsense mutations, and mutated mRNA is unstable; these mutants are recognized by a loss of the conductance of the Cl-channel in the affected epithelia. In class II, the protein is absent from the plasma membrane or present in small quantities. The mutations of this class represent the majority of CF alleles (DF508). The Class III conversions are situated in the ATP-binding domain. Class IV is recognized for changing the conduction of the Cl-channel. Classes V and VII depend on matured CFTR and mRNA function.13,14
CF is an autosomal genetic disease affecting the lungs and other organisms. In addition, it is the most commonly life-limiting monogenic disorder caused by the mutations of a gene encoding CFTR and is characterized by progressive lung destruction.2 CF is caused by several classes of mutations in the CFTR gene that cause CF. The CFTR is a cyclic AMP-activated chloride channel encoding a chloride ion channel content across epithelial cells. The deletion of phenylalanine residues at position 508 (F508del) is an essential mutation in CFTR, causing the degradation of the CFTR protein.3
Direct effects of CFTR dysfunction on disease
The dysregulation of the CFTR gene is related to immobile secretions, leading to inflammation that infiltrates neutrophils recruited by interleukin 8(IL-8). Therefore, this review study aimed to investigate the role of miRNAs as therapeutic and diagnostic biomarkers in CF and the relationship between these miRNAs and inflammation and the immune system. As mentioned earlier, CF is a genetic disorder that causes thick mucus to increase in organs, including the lungs and the pancreas. Thick mucus plugging and hyperhidrosis are direct effects of CFTR dysfunction. In the normal type, the mucus lining organs and body cavities (e.g., lungs and nose) is slippery and watery. However, in CF, thick mucus clogs the airways and makes breathing difficult. The mucus blocks the ducts in the pancreas and causes problems in digesting food by decreasing food absorption. The pathobiology of this matter is that after the CF gene, CFTR cloning, and protein production happened as a cAMP-regulated Cl− channel; in addition, causal mechanisms underlying the strong mucus phenotype of the disease became obscure. The mucus hyperproduction may not be a direct cause of a defective CFTR but can be a downstream consequence. In the lungs opposite, the other organs, the upregulation of mucin gene expression by inflammation results from chronic infection. Mucin glycoproteins (mucins) are a huge population of lung mucus that is heavily O-glycosylated and recognized by the MUC genes that encode their protein backbones. Many studies hypothesized that CFTR is directly involved in mucus production such as trafficking and mucin synthesis. Further, CFTR is indirectly involved in mucus obstruction. Several hypotheses showed the indirect effect of CFTR such as the dehydration hypothesis, innate immune hypothesis, and serous cell malfunction hypothesis. The dehydration hypothesis focusing on mutated CFTR fails in the imitation of making contributions to the desired ionic pressure because fabulous hydration over the mucus layer results in plump mucus, including concentrated mucins up to expectation collapses onto the underlying epithelium as much the first quadrant toward continual contamination then member disorder.
Indirect (or downstream) effects of CFTR dysfunction on disease
Unlike typical treatment plans with that amount of goal downstream outcomes concerning CFTR dysfunction, CFTR modulators aim at the imitation of unerring the underlying defect at the protein level. Traditional therapies as a target of downstream effects of CFTR dysfunction involve symptom management with clearances of airway infection treatment due to antimicrobials. The aim of the downstream subsequences of CFTR dysfunction is to correct CFTR deficiency at the protein level. This deficiency causes ion imbalance in the neutrophil and changes the degranulation patterns of neutrophils by increasing cytosolic chlorine and sodium and reducing magnesium. CFTR dysfunction decreases inflammation, improves microbial killing, and reduces the frequency of airway infection in CF. Inflammation is an early process in CF. CFTR dysfunction leads to a dysregulated inflammatory response. Before infection, CF airways are already in a proinflammatory process, causing tissue damage and chronic infection. By the way, CFTR dysfunction leads to leukocyte infiltration into the lung and intertwined tissue destruction. CF airways have an inflammatory environment recognized by the infiltration of inflammatory cells, primarily neutrophils, and a sticky mucus layer. Additionally, for restoring CFTR activity, CFTR modulators use anti-inflammatory factors by targeting intracellular processes such as ceramide accumulation, cytokine transcription, and macrophage-mediated phagocytosis of bacteria.
MiRNAs in cystic fibrosis
Initial reports
The first studies on the role of miRNAs in CF indicated that the levels of miRNAs have a relation with low expression of CFTR under hypoxic conditions.15 Some studies described the negative regulator effect of miR-200b on CFTR mRNA levels in human airway epithelia during hypoxia.
The results of another study represented that miR-126 is a target of Myb1 (TOM1) protein and regulates endosomal trafficking, also known as a negative regulator of IL-1 and lipopolysaccharide.
Direct miRNA binding to CFTR
MiRNAs, directly or indirectly, suppress the expression of CFTR. MiR101, miR-145, miR-223, miR-494, miR-509-3p, and miR-1246 directly target the 3′ UTR of CFTR and decrease its expression. The high expression of MiR-101 due to CS consequently decreases CFTR in the epithelial cells of the bronchus.16 The essential targets of the CFTR gene are miR-101, miR-494, miR-145, miR-124-5, miR-509-3p, and miR-223. In addition, miR-509-3p, miR-223, miR- 494, and miR-145 expressions were upregulated in CF.6 The role of miRNAs in CFTR expression is reined by the various information on phenotypic variability and lack of genotype-phenotype correlation among patients with the same mutation in the CFTR gene.17 The mechanism of miR-509-3P mediates polymorphisms in the 3′-UTR region of CFTR and decreases CFTR expression.
miRNA involvement with the other ion channels
MiR-138, as an indirect suppressing, downregulates CFTR with a repress of the SIN3A (switch-independent three-homolog A) transcription regulator.6,8,15 The upregulation of miR-9 expresses the activity of the gene anoctamin 1, a calcium-activated chloride channel in the epithelial cells of branches. Nonetheless, MiR-384, miR-494, and miR-1246 have the same effect on chloride channel regulation via the solute carrier family (SLC) 12 member 2 (SLC12A2) gene regulation. MiRNAs can also regulate the expression of NKCC1 (Na + -K + -2Cl--cotransport protein) or the sodium channel ENaC such as miR-183.10
miRNA alteration secondary to CF disease (hypoxia, infection, immunity, inflammation, and the like)
Numerous studies indicated that miRNAs affect innate immunity in CF and addressed miRNA’s relation with the Toll-like receptor (TLR) signaling pathway in CF. The Myb1 (TOM1) target is a Toll-interacting protein binding regulated by TLR2, TLR4, and IL-1β in CF bronchial epithelial cells. TOM1, which is downregulated by miR-126, negatively modulates TLRs. Recently studies demonstrated that the decreasing of miR-126 causes the hyporesponsive of TLR. Cathepsin S and caveolin 1/CAV1 are essential factors in CF fluid and the bridge between miRNAs and CF.18 miR-31 is a biomarker of CF and inhibits the transcription of cathepsin S by targeting interferon regulatory factor 1 (IRF1), which is part of the TLR signaling pathway as TLR2 can activate it. The activation of TLR2 and transcription of cathepsin S by IRF1 are common resident pathogens in CF lungs. According to recent studies, miR-31 is related to cathepsin S (CTSS) production via IRF-1 inhibition in CF epithelial cells.13 The overexpression of CTSS in CF patients increased the pulmonary neutrophilic infiltration of the lung, activated antimicrobial proteins, caused inflammation, and determined lung damage. Caveolin 1/CAV1, which is under expression with miR-199a-5P, is a negative regulator of TLR4 signaling due to PI3K-AKT signaling deviation, an indicator of CFTR-deficient macrophages. Studies confirmed an increase that the number of miRNAs implicated with TLR signaling in the CF.14 TLRs are innate immunity components and have a role in inflammation, thus they can act as a bridge between miRNAs and inflammation. The CF lung’s inflammation includes monocytes, macrophages, neutrophils, and dendritic cells. Studies examining miRNA expression in CF myeloid cells demonstrated the relation between miRNAs and myeloid cells in CF; for instance, miR-9 decreased, while miR-126 increased in neutrophils and mononuclear cells.13 MiR-17 and miR-93 are downregulated in CF bronchial epithelial cells. Decreasing miR-93 expression is correlated with IL-8 increasing. IL-8 is an essential chemokine in neutrophil function.19 The upregulation of IL-8 activity can cause the overexpression of miR-155, aggravating inflammation via p13/AKT activity in CF cell lines. MiR-155 overexpression occurs in lung epithelial cells and neutrophils in CF and downregulates the expression of the regulatory-associated protein of mTOR complex 1.17 miR-17 directly represses IL-8 as an anti-inflammatory factor. IL-8 production induces lipopolysaccharide and CPG- DNA through the TLR signaling pathway and leads to lung structural and cellular damage in CF patients.12 TLR signaling mediators affect epithelial cells and factors affecting the CFTR gene. The present review demonstrated that TOM1 is a vital gene suppressed by miR-126 and negatively regulates IL-1β with downregulating nuclear factor-κB (NF-kB) and mitogen-activated protein activity. Recent studies reported that miR-126 was reduced in CF patients’ lungs. The decrease of miR-126 is correlated with the upregulation of TOM1 and the downregulation of TLR2, TLR4, IL-1, IL-1β, tumour necrosis factor α (TNF-α), and NF-kB.2,5,18 Notably, a signaling molecule as small as miRNAs has severe potential when deregulated. Some studies represented that miR-215 modulates the cell cycle via the p-53 signaling pathway in CF patients. As a direct target of CFTR expression, miR-509-3p and miR-494 are increased in CF due to TNF-α and IL-1B exposure. The active macrophages of miR-146a increase to overproduce inflammatory cytokines such as IL-1B or IL-6 in response to infection. miR-146a plays a role in CF inflammation due to its negative impact on mucin 5AC (MUC5AC) production. MiR-145 is another factor contributing to CF-related inflammation with the downregulation of SMAD3, a negative modulator of the NF-kB-IL-8 pathway mediated by transforming growth factor beta (TGF-β). Phosphorylated (p) SMAD3 (pSMAD3) is an activator of TGF-β and decreases with miR-1343 in CF.3,5,20
Based on investigations, low CFTR proteins contribute to the upregulation of miR-101. miR-145 may increase due to the F508del mutation in CF patients. miR-145-5p and miR-223-3p have a function on the overexpression of F508del in CF. The evaluations revealed that miR-126 downregulation increases Myb1 (TOM1) expression in the CF lung.16 The high expression of TOM1 reduces inflammation in CF. Regarding the miR-145 function in CFTR, peptide nucleic acid (PNA) has a vital role in increasing the regulation of miR-145-5P. Therefore, PNAs target miR-145-5p by directly or indirectly binding miR-145-5p to the 3′-UTR CFTR mRNA sequences.12
MiRNAs as a therapeutics target in cystic fibrosis
miRNA biogenesis and function in animals
miRNA biogenesis begins in the nucleus, where the RNA-polymerase II-dependent transcription of a relatively large capped and polyadenylated transcript is known as primary miRNA (pri-miRNA). Pri-miRNA is processed by the RNase III endonuclease, Drosha, and its cofactor (Dgcr8) into smaller stem-looped structures known as precursor miRNAs (pre-miRNA). Pre-miRNAs are transported out of the nucleus by Exportin 5 into the cytosol, where further processing by a second RNase III enzyme (Dicer) leads to the generation of mature miRNA. The mature miRNA is associated with the miRNA-induced silencing complex (miRISC), where Watson-Crick base-pairing between the seed sequence of a mature miRNA and complementary sequences primarily located within 3’-UTRs of mRNAs results in post-transcriptional gene silencing.
Antisense oligonucleotide (ASO) binds to the complementary region in the target mature mRNA, forming a DNA or RNA hybrid, which then recruits the RNase-H enzyme that selectively cleaves the target mRNA sequence by hydrolyzing one of its internucleotide phosphate linkages and ablates the target gene expression. ASO physically blocks the binding motifs of the proteins of the cellular machinery, including the ribosome-binding site and translation initiation site in the mature mRNA. Furthermore, ASO can be designed to target pre-mRNA and interfere with RNA-processing events such as splicing or nuclear polyadenylation. The introduction of splice-switching AOs can therapeutically manipulate defective splicing by the steric blocking of the splicing motifs, where the splicing factors bind to the pre-mRNA, thus altering subsequent processes such as exon skipping, exon inclusion, and intron retention. Short interfering RNAs (siRNAs) are small double-stranded RNA molecules that have exact complementarity to a target mRNA. Once associated with the RISC complex, it binds to its target mRNA and induces gene silencing by preventing the translation of the mRNA. miRNA mimics are small double-stranded RNA molecules that are associated with and guide the RISC complex to its target mRNA. The mimic will bind with imperfect complementarity to its target mRNA, and translation will be blocked or the mRNA will be degraded, leading to gene silencing (Figure 1).
Oligotherapeutics as a field
ASOs as novel therapeutics have been identified by a nomenclature reported by Mann et al and are considered a potential way to develop the treatment of CF. Some studies demonstrated that oligonucleotides repair CFTR-encoded mRNA and cause a normal CFTR protein in patients with the Phe508del mutation. The mechanisms of this function are divided into RNase H-dependent and independent types. Independent mechanisms include the inhibition of ribosomal complex formation by inhibiting the translation and blocking of miRNA, as well as 5’ cap formation. ASOs target matured mRNA transcripts and missense mutations. Moreover, ASO can change the splicing and attenuate exon selection for the coverage of CFTR exon 10, increase polymorphisms, and cause CF. ASOs were used to amplify exon selection in cases where splicing mutations have led to exon skipping, including the T5 polymorphism in intron 9, resulting in CFTR exon 10 skipping. Additionally, it is the leading cause of CF. ASOs improve the process by acting as steric blockers and specialty bind to either splicing enhancers/silencer sequences. In addition, they produce non-functional mRNAs and decrease the nonsense-mediated pathway.
Recent studies revealed that miRNAs exist in extracellular conditions that can be detected in body fluids as disease biomarkers. Extracellular miRNA expressions were observed in several chronic lung diseases.18 Several studies highlighted the potential role of miRNAs in mediating CF pathophysiology related to TGF-β signaling. Numerous contents are involved in developing miRNA therapeutics.14 The critical way of miRNA suppression is using the oligonucleotide (antagomir), which binds to miRNA targets and promotes miRNA-mRNA connections. The overexpression of miRNAs was employed as a therapy to downregulate TGF-β pathway members.13 Depending on miRNA functions in disease, the miRNA mimics increase or decrease the expression via suppressing miRNA action (anti-miRs). According to respiratory-based delivery systems, miRagen therapeutics is a trial for miR-29 mimic (MRG-201) in pathologic fibrosis.19 As potential predictive biomarkers, miR-155 high expression serum levels have been detected in the airway cells of CF patients. Single nucleotide polymorphisms demonstrated phenotype variability in the 3’UTR of the miRNA-targeted genes of CFTR and could act as a mutation. The CFTR 3’UTR polymorphisms play an important role in the heterogeneous phenotype, and the molecular analysis of the 3’UTR of CFTR is recognized as a diagnostic tool in CF. Endogenous miRNAs affect the expression of CFTR with the therapeutic strategy of miRNA mimics and antimiR agents. miR-138 indirectly downregulates SIN3A with the impairs of CFTR. The management of miR-138 expression resets the CFTR redundancy via up-regulating SIN3A. PNAs and target site blockers (TSBs) are the other strategies of the miRNA therapeutic target.12,17
Another disease process using miRNA manipulation
PNA as DNA analog and miRNA target protectors in nucleic acid is considered a valid anti-miR strategy. PNAs can be used to target miR-145, miR-509-3p, and miR-101-3p in CFTR expression and then increase this expression via the interaction of miRNAs with mRNA targets. TBS are oligonucleotides that affect some marks. TBS can competitively bind to a miRNA target site in mRNA.18 The CFTR expression increased due to TBS targeting miR-101 and miR-145 in the CFTR 3′ UTR. Recent studies have suggested the inhibition of CFTR mediated by miRNAs via CFTR-specific TSBs in CF bronchial epithelial cells. The bottom line in miRNAs has an essential role in the therapeutic biomarker and diagnosis of CFTR. As mentioned earlier, miR-145 expression increased due to TGF-β upregulation and caused CFTR inhibition. Thus, an antimiR strategy directed against miR-145 can enhance the F508del CFTR correction in CF airway epithelia.8,12
Previous trials of miRNA manipulation to improve CFTR function or response to therapy
Our previous study on miRNA manipulation to improve CFTR correction showed that miR-145 overexpression reduced CFTR protein synthesis and function. MiR-145 directly binds to the 3′-UTR of the human CFTR transcript. In addition, the results demonstrated that the CFTR knockdown increases endoglin and TGF-β in airway epithelia. Further, the loss of CFTR by siRNA knockdown or functional CFTRinh‐172 inhibition increases endoglin and TGF-β signaling. A recent study represented that the downregulation of CFTR expression during hypoxia is HIF-1-dependent and relies on the 5′ and 3′ UTRs of CFTR mRNA. The study on the MiR200 effect on CFTR showed that miR-200b is induced by hypoxia in a HIF-1-dependent manner in human lung epithelial cells. Based on the result of this study, the increase in miR-200b levels was negatively correlated with the respective decrease in CFTR mRNA and protein and supported the role of miR-200b in regulating CFTR expression. Similarly, our previous studies indicated that the loss of CFTR function increased endoglin expression with the corresponding increase in TGF‐β1 transcription and signaling.
Oligotherapeutics to address the genetic modifiers of CF disease (TGF-β and probably others)
TGF-β
TGF-β has been identified as a modulator of disease phenotypes. TGF-β polymorphisms such as C-509T polymorphism in the promoter region and T29C polymorphism in codon 10 are a mechanism of lung pathology in CFTR. Canonical TGF-β signaling acts via the phosphorylation of SMAD transcription factors such as smad2 and smad3. The TGF-β signaling alteration of miRNA transcription may depend on non-Smad molecules such as ERKs, JNK/p38, RhoA, and PI3K/Akt. Multiple studies indicated that TGF-β and miRNAs have a relationship in CF; for example, miRNA-145 mediates the TGF-β-induced suppression of CFTR. Additionally, alterations in TGF-β binding miRNA may regulate this genetic modifier of CF disease progression. Based on the results of our previous study, miR-145 could manage the TGF-β inhibition of CFTR synthesis and function in epithelial cells. In this study, the binding of miRNA-145 to the CFTR 3′-UTR has been reported by our assays. For example, miR-200, miR20, miR-1343, and miR-21 directly target TGF-beta receptors or downstream signaling. The results of a recent study showed that miR-200b decreases the expression of CFTR mRNA during normoxia and hypoxia. Multiple other miRNAs (other examples can be similarly provided) alter the immune phenotype, transitioning to a Th2 or Th17 physiology.
Targeting the other genetic modifiers
Recent studies have suggested that the other candidate gene studies were involved in CF disease. In this regard, the most important candidates are nitric oxide synthase (NOS), glutathione and glutathione-S-transferase,mannose-binding lectin 2,cell surface receptors, macrophage migration inhibitory factor, ion channels, mucinex, and cytokines. Three isoforms of NOS, including neuronal NOS (NOS1), inducible NOS (NOS2), and endothelial NOS (NOS3) are involved in the NOS group. NOS1 is an important neurotransmitter that controls the bronchomotor. Bronchial epithelial cells and macrophages express NOS2 in the lung. The exhaled NO decrease in CF patients. In patients with F508 dell mutation, both nasal and bronchial NO decrease during exhalation.
Augmenting CFTR-directed therapeutics
Factors enhancing the intracellular concentration of cAMP and amplification of PKA activity and the R domain phosphorylation level increase the activity of deficient CFTR. Drumm et al found that IBMX (3-isobutyl-1-methylxanthine) inhibits phosphodiesterase and enhances intracellular cAMP and the response of F508del-CFTR to activation. Most studies reported that genistein is a tyrosine kinase inhibitor that increases intracellular cAMP and then activates CFTR. VX-770 (Ivacaftor) is another CFTR activator that enhances the activity of F508del – CFTR in bronchial epithelial cells. VX-770 activates the hydrolysis of ATP and NBD domain dimerization. As a new therapy, Ivacaftor is the proof-of-concept of CFTR modulation which can supplement various CFTR mutants. Ivacaftor was first tested in cells and upregulated chloride secretion and active CFTR. As described in our present review, Phe508del is the most common mutation in CF, and studies demonstrated that Ivacaftor monotherapy in the homozygous and heterozygous Phe508del population is useful. Further, investigations revealed that Ivacaftor improves channel open and chloride ion conductance in the epithelial cells of CF patients.
Currently available therapy (our miR-145 Work)
Currently, our study results on miR-145 mediating TGF-β inhibition of CFTR synthesis and function in airway epithelia represented that miR-145 manages the TGF-β inhibition of CFTR synthesis and function in airway epithelia. Specific antagonists to miR-145 inhibit TGF-β signaling to restore F508del CFTR modulation. In addition, miR-145 antagonism may offer a novel therapeutic opportunity to enhance the therapeutic benefit of F508del CFTR correction in CF epithelia. To the best of our knowledge, this is the first study that demonstrated that miR-145 is a targetable mechanism linking to TGF-β signaling to reduce CFTR function with CFTR modulator strategies.
Conclusion
miRNAs play a critical role in gene regulation and disease pathogenesis. This review confirmed the contribution of miRNA to CFTR gene expression, CF disease pathogenesis, and oligothereutic opportunities. Targeting specific miRNA has great potential in the treatment of CF. Briefly, molecular target therapy has an important effect on target cells which can reduce tissue damage.
Authors’ Contribution
Conceptualization: Delaram Mehrzadeh, Mahsa Shorodi.
Methodology: Delaram Mehrzadeh, Mahsa Shorodi, Sina Rahimpour.
Project Administration: Sina Rahimpour.
Resources: Mahsa Shorodi.
Validation: Sina Rahimpour.
Writing — Original Draft: Mahsa Shourudi.
Writing — Review & Editing: Sina Rahimpour.
Supervision: Sina Rahimpour.
Competing Interests
None.
Ethical Approval
Not applicable.
References
- Cook NL, Pereira TN, Lewindon PJ, Shepherd RW, Ramm GA. Circulating microRNAs as noninvasive diagnostic biomarkers of liver disease in children with cystic fibrosis. J Pediatr Gastroenterol Nutr 2015; 60(2):247-54. doi: 10.1097/mpg.0000000000000600 [Crossref] [ Google Scholar]
- Calvopina DA, Chatfield MD, Weis A, Coleman MA, Fernandez-Rojo MA, Noble C. MicroRNA sequencing identifies a serum microRNA panel, which combined with aspartate aminotransferase to platelet ratio index can detect and monitor liver disease in pediatric cystic fibrosis. Hepatology 2018; 68(6):2301-16. doi: 10.1002/hep.30156 [Crossref] [ Google Scholar]
- Choi MR. Commentary for the article: microRNA-1246 regulates proliferation, invasion and differentiation in human vascular smooth muscle cell by targeting cystic fibrosis transmembrane conductance regulator (CFTR). Pflugers Arch 2021; 473(2):135-7. doi: 10.1007/s00424-021-02516-3 [Crossref] [ Google Scholar]
- De Palma FDE, Raia V, Kroemer G, Maiuri MC. The multifaceted roles of microRNAs in cystic fibrosis. Diagnostics (Basel) 2020; 10(12):1102. doi: 10.3390/diagnostics10121102 [Crossref] [ Google Scholar]
- Fabbri E, Tamanini A, Jakova T, Gasparello J, Manicardi A, Corradini R. Treatment of human airway epithelial Calu-3 cells with a peptide-nucleic acid (PNA) targeting the microRNA miR-101-3p is associated with increased expression of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Eur J Med Chem 2021; 209:112876. doi: 10.1016/j.ejmech.2020.112876 [Crossref] [ Google Scholar]
- Fabbri E, Tamanini A, Jakova T, Gasparello J, Manicardi A, Corradini R. A peptide nucleic acid against microRNA miR-145-5p enhances the expression of the cystic fibrosis transmembrane conductance regulator (CFTR) in Calu-3 cells. Molecules 2017; 23(1):71. doi: 10.3390/molecules23010071 [Crossref] [ Google Scholar]
- Fesen K, Silveyra P, Fuentes N, Nicoleau M, Rivera L, Kitch D. The role of microRNAs in chronic pseudomonas lung infection in cystic fibrosis. Respir Med 2019; 151:133-8. doi: 10.1016/j.rmed.2019.04.012 [Crossref] [ Google Scholar]
- Glasgow AMA, De Santi C, Greene CM. Non-coding RNA in cystic fibrosis. Biochem Soc Trans 2018; 46(3):619-30. doi: 10.1042/bst20170469 [Crossref] [ Google Scholar]
- Lutful Kabir F, Ambalavanan N, Liu G, Li P, Solomon GM, Lal CV. MicroRNA-145 antagonism reverses TGF-β inhibition of F508del CFTR correction in airway epithelia. Am J Respir Crit Care Med 2018; 197(5):632-43. doi: 10.1164/rccm.201704-0732OC [Crossref] [ Google Scholar]
- McKiernan PJ, Greene CM. MicroRNA dysregulation in cystic fibrosis. Mediators Inflamm 2015; 2015:529642. doi: 10.1155/2015/529642 [Crossref] [ Google Scholar]
- McKiernan PJ, Molloy KP, Cryan SA, McElvaney NG, Greene CM. X chromosome-encoded microRNAs are functionally increased in cystic fibrosis monocytes. Am J Respir Crit Care Med 2018; 197(5):668-70. doi: 10.1164/rccm.201707-1417LE [Crossref] [ Google Scholar]
- Pan D, Liu G, Li B, Jiang J, Chen W, Li W. MicroRNA-1246 regulates proliferation, invasion, and differentiation in human vascular smooth muscle cells by targeting cystic fibrosis transmembrane conductance regulator (CFTR). Pflugers Arch 2021; 473(2):231-40. doi: 10.1007/s00424-020-02498-8 [Crossref] [ Google Scholar]
- Sonneville F, Ruffin M, Coraux C, Rousselet N, Le Rouzic P, Blouquit-Laye S. MicroRNA-9 downregulates the ANO1 chloride channel and contributes to cystic fibrosis lung pathology. Nat Commun 2017; 8(1):710. doi: 10.1038/s41467-017-00813-z [Crossref] [ Google Scholar]
- Sonneville F, Ruffin M, Guillot L, Rousselet N, Le Rouzic P, Corvol H. New insights about miRNAs in cystic fibrosis. Am J Pathol 2015; 185(4):897-908. doi: 10.1016/j.ajpath.2014.12.022 [Crossref] [ Google Scholar]
- Mooney C, McKiernan PJ, Raoof R, Henshall DC, Linnane B, McNally P. Plasma microRNA levels in male and female children with cystic fibrosis. Sci Rep 2020; 10(1):1141. doi: 10.1038/s41598-020-57964-1 [Crossref] [ Google Scholar]
- Nicola T, Kabir FL, Coric T, Wall SB, Zhang W, James M. CFTR dysfunction increases endoglin and TGF-β signaling in airway epithelia. Physiol Rep 2019; 7(4):e13977. doi: 10.14814/phy2.13977 [Crossref] [ Google Scholar]
- Pierdomenico AM, Patruno S, Codagnone M, Simiele F, Mari VC, Plebani R. MicroRNA-181b is increased in cystic fibrosis cells and impairs lipoxin A(4) receptor-dependent mechanisms of inflammation resolution and antimicrobial defense. Sci Rep 2017; 7(1):13519. doi: 10.1038/s41598-017-14055-y [Crossref] [ Google Scholar]
- Vencken SF, Greene CM. Toll-like receptors in cystic fibrosis: impact of dysfunctional microRNA on innate immune responses in the cystic fibrosis lung. J Innate Immun 2016; 8(6):541-9. doi: 10.1159/000444687 [Crossref] [ Google Scholar]
- De Santi C, Greene CM. Challenges facing microRNA therapeutics for cystic fibrosis lung disease. Epigenomics 2020; 12(3):179-81. doi: 10.2217/epi-2019-0395 [Crossref] [ Google Scholar]
- Noel S, Leal T. Emerging roles of microRNAs in cystic fibrosis—from pathogenesis to development of new therapies. In: Wat D, ed. Cystic Fibrosis in the Light of New Research. IntechOpen; 2015. 10.5772/60699.